
Dementias Caused by Persistent Pathogens and the Protective Role of HLA Against them
Lisa M. James1,2,3, Apostolos P. Georgopoulos1,2,3,4*
1Brain Sciences Center, Department of Veterans Affairs Health Care System, Minneapolis, USA
2Department of Neuroscience, University of Minnesota Medical School, Minneapolis, USA
3Department of Psychiatry, University of Minnesota Medical School, Minneapolis, USA
4Department of Neurology, University of Minnesota Medical School, Minneapolis, USA
Abstract
Dementia is a leading cause of death worldwide, representing a significant global burden. In addition to genetic and lifestyle factors that have been widely linked to dementia, pathogens are increasingly recognized as contributing to the development of dementia. Here we discuss the role of human leukocyte antigens (HLA) in maintaining brain health by facilitating the elimination of pathogens and highlight evidence suggesting that the inability to eliminate pathogens contributes to dementia. Finally, we briefly review common forms of dementia including Alzheimer’s disease, vascular dementia, frontotemporal dementia, Lewy body dementia, and prion dementia in an effort to contextualize the role of persistent pathogens across the various dementia phenotypes.
Introduction
An estimated 50 million people are living with dementia worldwide, and that number is expected to triple by 20501. Dementia is a broad term that refers to conditions reflecting a decline in cognitive, behavioral, motor, and/or social functioning that eventually interferes with the ability to live independently. The most commonly recognized forms of dementia include Alzheimer’s disease (AD), vascular dementia, Lewy body dementias, and frontotemporal dementia. Prions and prion-like mechanisms associated with other neurodegenerative disorders are also implicated in dementia. The causal factors and mechanisms underlying brain deterioration associated with dementia as well as less pernicious forms of age-related cognitive decline have long been intensely investigated; however, there is a burgeoning emphasis on also understanding factors that promote the maintenance of healthy brain functioning across the lifespan. Here we will briefly review the emerging picture of immunogenetic mechanisms underlying brain health and disease centering around human leukocyte antigens (HLA) and discuss the implications of this immunogenetic influence in terms of common forms of dementia.
Healthy Brain Aging
It is widely accepted that the trajectory of cognitive functioning across the lifespan is influenced by both lifestyle factors such as diet and exercise as well as genetics. Among genetic influences, apolipoprotein E is well-established in playing a role in the maintenance of cognitive function and decline, depending on genotype. Specifically, the apolipoprotein E4 allele has been shown to be associated with brain atrophy2, neural network dysfunction3,4, increased risk for AD5,6, and reduced cognitive performance among cognitively healthy individuals7. Conversely, the apoE2 alleles have been linked to preserved cognitive functioning and reduced risk of AD8.
HLA neuroprotection
Emerging evidence suggests that HLA may also play a crucial role in brain health and disease. HLA genes encode for cell surface glycoproteins that are involved in immune surveillance and elimination of foreign antigens such as viruses and bacteria via the facilitation of cell destruction (Class I) or antibody production (Class II). These processes, however, hinge on a match between HLA molecules and antigenic peptides as a first crucial step (Fig. 1). The HLA genes are the most highly polymorphic in the human genome, an evolutionary consequence aimed at maximizing host protection from foreign antigens/pathogens. However, each individual has a limited repertoire of HLA molecules, restricting the scope of foreign antigens that can be successfully eliminated. Furthermore, HLA molecules differ in their binding affinity and ability to mount an immune response. Among the tens of thousands of HLA alleles, some occur at much higher frequencies in a population than others owing to their relative advantage in host protection via antigen elimination. For example, HLA DRB1*13 alleles have been shown to exert broad protective effects against several conditions affecting the brain, including various autoimmune disorders9,10 and Gulf War Illness, a neuroimmune condition related to military service during the 1990-91 Persian Gulf conflict11,12. With regard to Gulf War Illness, we found that HLA DRB1*13:02 protected against brain atrophy13 which is typical of those with the condition14
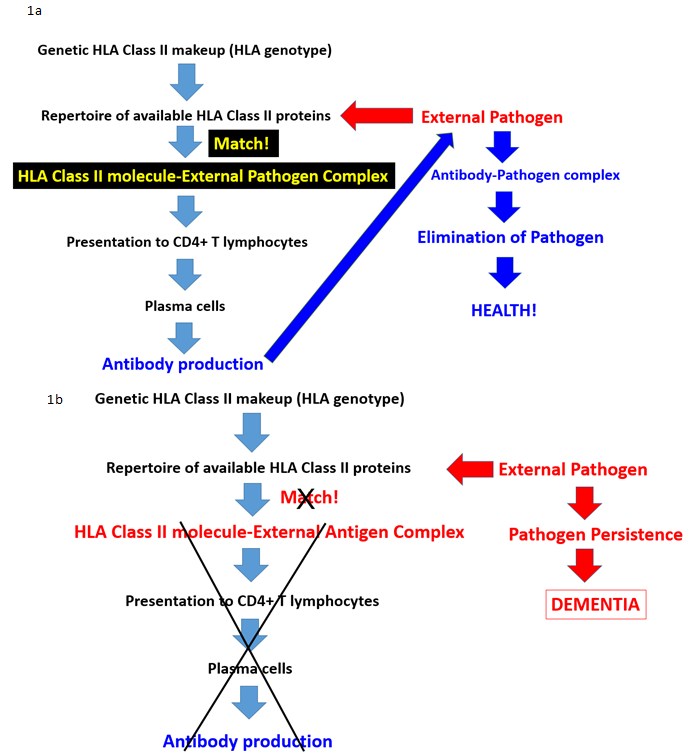
Figure 1. Schematic diagram illustrating the steps of antibody production in health (Fig. 1a) and, conversely, the disruption of antibody production at various possible stages leading to persistent pathogens and disease (Fig. 1b). Adapted from James et al., 2017.13
Brain atrophy is also viewed as a consequence of aging, with typical rates of atrophy approaching 5% per decade15. Given the neuroprotective effects observed in other conditions, we investigated potential protective effects of HLA DRB1*13 against age-related brain changes. Again we found that HLA DRB1*13 alleles (HLA-DRB1*13:02 primarily but also HLA-DRB1*13:01 to a lesser extent) protect against age-related brain atrophy16 and function17, even in the presence of apoE4. The role of HLA in pathogen elimination, in conjunction with evidence of HLA-DRB1*13 protection against age-related brain changes, led us to speculate that these alleles may protect against dementia via the elimination of pathogens. Indeed, in a recent genetic epidemiological study, we demonstrated that dementia prevalence (any type of dementia) in 14 Continental Western European countries is inversely associated with the population frequency of HLA DRB1*13:02 and was not accounted for by apoE418 (Fig. 2a). In fact, as the population frequency of HLA DRB1*13:02 doubled, the prevalence of dementia was reduced by one-third (Fig. 2b). Remarkably, the frequency of HLA DRB1*13:02 was shown to account for 45.2% of the variance in dementia rates. Given the role of HLA Class II alleles in elimination of foreign antigens by the production of specific antibodies against them, these findings suggest that individuals who possess the DRB1*13:02 allele, as well as populations in which the allele is common, have enhanced protection against dementia conferred by the successful elimination of the foreign pathogens. In the absence of such successful elimination, the pathogens may persist causing inflammation and cell damage19, the effects of which are likely to be exacerbated in apoE4 carriers due to the neurotoxicity of apoe4 fragments relative to other isoforms20,21. Thus, we have hypothesized that any foreign pathogen that cannot be successfully eliminated due to HLA-antigen incongruence may be causally involved in dementia development20 (Fig. 1b). Unlike theories that are restricted to reactivation of latent infection as causing dementia, we proposed that the persistence of foreign pathogens, even in the absence of frank infection, may cause gradual damage to the brain that results in dementia20.
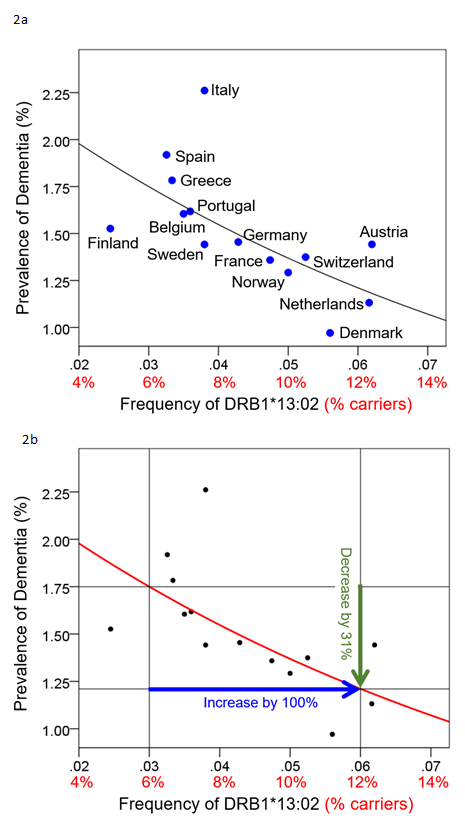
Figure 2. Dementia prevalence is plotted against DRB1*13:02 frequency for 14 Continental Western European countries (Fig 2a) and the quantitative effect whereby an increase of 100% (doubling) of the DRB1*13:02 frequency results in 31% reduction in dementia prevalence is illustrated. Reprinted from James & Georgopoulos 2019.18
Pathogens and Dementia
Once considered controversial, the role of pathogens in dementia is increasingly recognized and investigated. Indeed, numerous pathogens have been implicated in AD, including, most prominently, herpes viruses in addition to bacterial spirochetes such as those related to gum disease, among others22,23. These infections have not only been shown to increase the risk of AD but the role of microbes in AD is also supported by evidence of the presence of microbes in brain tissue, particularly in regions that are impacted in AD, and co-localization of those microbes with typical AD pathology22,23. A recent study of mice intranasally infected with the H5N1 virus provides a link between pathogens and Lewy body dementias (LBD)24. Specifically, the authors demonstrated that the H5NI virus enters the CNS via cranial nerves, affecting the brainstem first before spreading to the midbrain and cortex and that the virus can infect neurons by peripheral infection and by hematogenous spread subsequent to release from dying cells. Furthermore, they demonstrated aggregation of alpha-synuclein that co-localized with brain regions showing evidence of viral infection, including the hippocampus, cortex, and brainstem, similar to that seen in human proteinopathies. Finally, the authors reported evidence of microgliosis for the entire 90-day study period in spite of evidence that the acute infection had resolved by day 21, suggesting a prolonged inflammatory response resulting from the initial infection as might be expected with persistent pathogens. Prolonged inflammation has been linked to nearly all types of dementia25-27. Furthermore, because brain regions that are critical to cognitive processes including memory, attention, emotion, and perception contain more enzymes involves in inflammatory responses than do other regions of the brain they are thought to be at increased risk of cumulative damage from even low-level chronic inflammation28.
The specific dementia phenotype may be dependent on the type of pathogen and its mode of entry into the brain. For example, microbial pathogens may cross the blood-brain-barrier and/or blood-cerebrospinal fluid barrier via one or more of several mechanisms, including transcellular penetration, paracellular entry, or by infected leukocytes circulating peripherally29; they may also access the central nervous system through cranial nerves including most prominently the olfactory and trigeminal nerves before migrating to higher brain regions including those that have been implicated in dementia29. Access to the CNS through the olfactory and trigeminal nerves has been shown to be highly relevant to human herpesviruses as well as several types of influenza A29. Furthermore, researchers have demonstrated evidence of apoptosis in olfactory sensory neurons of virally-infected hosts which were suggested to be aimed at host protection from microbial invasion through the nasal cavity30. Indeed, olfactory dysfunction resulting in loss of smell has been implicated in nearly every type of dementia31. Taken together, compelling evidence supports the role of pathogens in dementia. Consistent with the mounting literature implicating pathogens in dementia and the immunogenetic (i.e., HLA) influence on pathogen elimination or, conversely, the persistence as described above, we have proposed the concept of persistent pathogen dementia20 which we characterize below.
Persistent Pathogen Dementia (PPD)
As noted above, several pathogens have been linked to dementia (primarily AD) including herpes simplex virus type 1 (HSV-1), Helicobacter pylori (h. pylori), Chlamydophila pneumonia, and Borrelia burgdorferi among several others23. Perhaps more than any other microbe, HSV-1 has been robustly associated with AD. The prevailing theory suggests that reactivation of latent HSV-1 infection in apoE4 carriers contributes to AD. The percent of dementia cases attributable to pathogens is unknown; however, evidence of HSV1 in the brains of apoE4 carriers has been shown in nearly 60% of cases32, suggesting high rates of pathogen-linked dementia. In addition to current theories implicating reactivation of latent infections, we hypothesize that fragments of pathogens that elude elimination due to HLA-antigen incongruence will similarly affect the brain20. To that end, any pathogen that is not able to be eliminated may contribute to PPD.
Persistent Pathogen Dementia (PPD)
The primary risk factor associated with PPD is exposure to pathogens (e.g., viruses, bacteria); however, rates of pathogen exposure do not square with rates of dementia indicating that pathogen exposure alone is not sufficient to produce dementia. For example, the majority of U.S. adults age 30 and older have been exposed to HSV-133 yet the U.S. population prevalence of dementia is less than 2%34, highlighting the role of additional factors. As with other dementias, apoE4 is considered a risk factor for PPDs. In the case of HSV-1, it has been suggested that HSV in apoE4 carriers is associated with high rates of dementia whereas neither apoE4 nor HSV1 alone raise risk32. An additional risk-factor appears to be a lack of immunogenetic (i.e., HLA) protection18. Because each HLA molecule matches specific epitopes, each person’s ability to eliminate pathogens is determined by their HLA profile as discussed above. Further, because each individual has a limited number of HLA genes that, in turn, determine which pathogens can be successfully eliminated, some individuals may lack protection against certain pathogens due to HLA-antigen incongruence.
Neuropathology
There is some evidence that neuropathology associated with persistent pathogens parallels and is associated with well-established dementia pathology. For example, H5N1 influenza has been shown to increase alpha-synuclein in pre-clinical studies24. Similarly, HSV-1 has been shown to increase AD-related amyloid-beta (Aβ)35 and tau36 in cell cultures and to co-localize with amyloid plaques in the human brain37. Similarly, HIV has been shown to modulate the production of Aβ and tau38. These findings dovetail with other lines of research demonstrating antimicrobial effects of Aβ39,40. That is, Aβ is thought to play a crucial role in the brain’s immune system by trapping pathogens. Similarly, there is evidence that tau accumulation is a neuroprotective response to oxidative stress, leading to the conclusion that “inclusion formation represents adaptation, or productive, beneficial response to the otherwise neurodegenerative process”41. Thus, from the PPD perspective, in the absence of an HLA-antigen match, persistent foreign antigens promote the production of plaques and tangles in an effort to protect the brain from foreign invaders. Notably, such protective effects do not preclude the possibility that accumulation of Aβ and tau tangles eventually becomes toxic; however, this suggests that exposure to pathogens, in the absence of protective HLA that can eliminate them, plays a fundamental role in dementia.
Neuroimaging
Neuroimaging findings related to PPD are likely to be as varied as the underlying pathogens. Indeed, each of the herpes viruses has been associated with unique neuroimaging features, with reactivation following the same patterns as initial infection42. Similarly, dementia related to HIV has been shown to be distinct from typical AD43. Characteristic neuroimaging markers for various persistent pathogens may be identified in future studies.
Treatment
Effective interventions for PPD are likely to center around the elimination of pathogens. To that end, recent studies of anti-herpes antivirals have shown promise in reducing the incidence of dementia among individuals with herpes zoster44 and HSV145 in two large-scale studies. If research continues to support the hypotheses that Aβ and tau accumulation provide neuroprotective functions, and antimicrobial functions, in particular, treatments aimed at modulating Aβ and tau may not be effective; instead, if amyloid and tau are stimulated in order to protect the brain from pathogens, elimination of the pathogen is called for.
Summary
Thus far we have shown that immunogenetic protection conferred by HLA genes guards against neurodegeneration associated with neuroimmune conditions and age-related processes at the individual level, and protects against dementia at the population level via facilitating the elimination of pathogens. For those lacking immunogenetic protection, exposure to HLA-incongruent pathogens may persist, causing inflammation and neurodegeneration that may result in dementia. We have also shown that pathogens are increasingly recognized as playing a prominent role in dementia and provided an overview of characteristics proposed to be associated with PPD. Next, we provide a brief overview of commonly recognized types of dementia, noting evidence suggesting the role of pathogens in where applicable. We hypothesize that all dementias below, except for vascular dementia, are indeed subsets of PPD.
Alzheimer’s Dementia
AD is the most common cause of dementia, accounting for 60-80% of dementia cases. Although AD may be diagnosed before age 65, the vast majority of cases are seen in those 65 and above; the more typical late-onset type is the focus here. Memory impairments are the hallmark symptom of AD; however, impairments in judgment and social functioning, changes in mood and personality, and motor deficits are also typical. Because AD is a progressive disease, the level of dysfunction increases over time such that at the final stages, affected individuals are completely impaired and unable to perform basic life functions.
AD Risk factors
A number of risk factors have been associated with dementia. Among the non-modifiable risk factors are age and genetics. After age 65, the risk for dementia increases with age such that one-third of people aged 85 or older are diagnosed with dementia46. A number of genes have been implicated in AD47. Among genetic risk factors, the apolipoprotein E4 (apoE4) allele has the most robust association with AD with risk increasing in a dose-dependent manner according to the number of apoE4 alleles5. ApoE is involved in a number of functions including cholesterol transport and metabolism, neuroplasticity, and neuronal repair21. Relative to apoE2 and E3, the E4 allele is unstable and its expression results in neurotoxic fragments that ultimately contribute to neurdogeneration20,21. However, apoe4 is neither necessary nor sufficient to produce AD; recent studies have identified various other genes that appear to be related to AD including several associated with immune response and inflammation6,47. In addition to nom-modifiable risk factors, a number of lifestyle-related factors have been implicated in dementia including diet, physical and mental exercise, diabetes, obesity, and smoking. Evidence suggests that a reduction in modifiable risk factors would lead to a significant decline in dementia incidence48. The recent decline in dementia incidence in many countries may be attributable to reduced risk owing to changes in modifiable risk factors. Finally, as previously noted, numerous pathogens have been implicated as a risk factor for AD22,23.
AD Neuropathology
The hallmark signs of AD are extracellular Aβ plaques and intracellular neurofibrillary tau tangles. Amyloids are protein aggregates that stick together forming fibrils. In the case of AD, it is believed that misfolded Aβ oligomers accumulate, forming plaques that disrupt synapses and ultimately contribute to neurodegeneration49. Protein misfolding and aggregation has also been implicated in Parkinson’s disease and prion diseases, both of which are also associated with dementia49; indeed, it has been suggested that Aβ and tau are themselves prion-like in that they may slowly self-propagate, ultimately contributing to widespread neurodegeneration50-52. Despite considerable evidence linking Aβ to dementia, mounting research has called into question the central role of Aβ plaques in AD53. First, postmortem autopsy of brain specimens54,55 and in vivo positron emission tomography56 have revealed evidence of significant amyloid plaque burden in the absence of memory impairment. Second, as summarized elsewhere53, several therapeutic strategies aimed at clearing Aβ from the brain have failed and, in some cases, produced severe side effects. Finally, Aβ has been shown to have important physiological functions including anti-microbial activity39,57,58. That is, accumulation of Aβ may result from immune-mediated mechanisms aimed at trapping pathogens, the persistence of which could facilitate the accumulation of Aβ. While it is recognized that Aβ likely participates in dementia, there are growing calls for the scientific community to “devote its utmost efforts to identify the real culprit of AD53”, which we have proposed is persistent pathogens20.
Tau is a highly soluble protein involved in microtubule stabilization, primarily in axons, that has also been implicated in AD. In the AD brain, tau is hyperphosphorylated relative to healthy tau59. Hyperphosphorylated tau becomes insoluble and misfolded protein aggregates form neurofibrillary tangles that accumulate in cells leading to impairment in several downstream processes including mitochondrial and synaptic function and axonal transport, and ultimately cell death39. Tau has also been implicated in AD-associated neuroinflammation60 and has been shown to interact with insulin to modulate AD61. Tau tangles are more correlated with cognitive impairment and AD severity than Aβ62,63, leading many to consider tau as fundamentally underlying AD. However, others have noted evidence of tau pathology in prepubertal human brains64 and in cognitively healthy individuals65, and have shown that neurons with neurofibrillary tangles can live for decades66, raising criticism for the hypothesis that tau mutations drive AD. Furthermore, as with Aβ, some have suggested that phosphorylated tau (and other intracellular inclusions) serve a neuroprotective role against adverse conditions such as oxidative stress and/or toxin exposure67. In this scenario, in which hyperphosphorylation of tau is secondary to other local cellular environmental insults, the long-term consequences may be deleterious (including neuronal death) albeit protracted. Finally, if tau indeed serves a protective role, AD interventions aimed at reducing tau pathology may be counterproductive.
AD Neuroimaging
Considerable effort is being made to detect AD in vivo with diagnostic neuroimaging moving along several fronts. Structural MRI is one of the most widely used neuroimaging techniques in the diagnosis of AD. Decades of research have converged to establish a signature of AD-related brain atrophy characterized by initial effects on the medial temporal lobe (i.e., entorhinal cortex and hippocampus), followed by an extension to the rest of the cortex, with the motor areas spared until later in the disease process, as reviewed elsewhere68. In addition, several subcortical structures including the amygdala, basal ganglia, caudate, and putamen, among others have been shown to exhibit significant atrophy in patients with AD68. Advances in neuroimaging have helped identify several subtypes of AD that are atypical relative to the AD signature described above. Indeed, mounting evidence suggests there is substantial heterogeneity in patterns of brain atrophy associated with AD with several subtypes following atypical patterns including one that spares the hippocampus, one that predominantly involves limbic structures, and one that is characterized by minimal atrophy69-70. Although speculative, it is possible that the heterogeneity observed in AD is related to the varied mechanisms of pathogen entry into the brain as described previously29.
Positron emission tomography (PET) is also widely utilized in clinical evaluations of AD. Amyloid, tau, and 18fluorodeoxyglucose (FDG) PET tracers have been used to noninvasively visualize amyloid and tau deposition and glucose consumption in the brain, respectively, each providing novel and complementary indications of AD-related pathological processes71. PET is also being used for in vivo detection of neuroinflammation as evidenced by microglia activation and reactive astrocytosis71. Increasingly, clinical scientists are advocating for the use of multimodal neuroimaging to detect the variety of pathological brain changes associated with AD, and ultimately increase diagnostic accuracy.
AD Treatment
To date, there is no cure for AD and treatment is aimed at managing symptoms as reviewed elsewhere72. Briefly, cholinesterase inhibitors and Memantine, an NMDA receptor agonist, have been licensed for AD with some benefit observed for cognition and mood symptoms. Neuropsychiatric symptoms in AD are commonly treated with atypical antipsychotics, antidepressants, or anticonvulsants with modest benefits and, in some cases, the potential for significant side effects. Finally, several disease-modifying treatments are under investigation, many of which are aimed at targeting amyloid-beta and tau; none have shown conclusive efficacy for AD yet. Given the lack of effective treatments for AD, the importance of AD prevention through lifestyle interventions such as exercise and a healthy diet has gained prominence.
Lewy Body Dementia and Parkinsonian Dementia
Lewy Body dementia (LBD), which include dementia with Lewy bodies and Parkinson’s disease dementia, are the second most common type of dementia in older adults, accounting for up to 30% of dementia cases. Men are disproportionately affected by LBD73. Clinically, LBDs are difficult to distinguish from AD in that both are characterized by a progressive decline in cognitive function, however, LBD is characterized by periods of normal cognition alternating with a cognitive impairment which is not typical of AD. Individuals with LBD are also prone to hallucinations and parkinsonian motor symptoms, and rapid eye movement sleep behavior disorders are common and have been shown to be predictive of LBD74.
LBD Neuropathology
As with several other forms of dementia, misfolded aggregated proteins are associated with LBD. The hallmark of LBD is alpha-synuclein neuronal inclusions. Alpha-synuclein is a protein found predominantly in the brain (e.g., neocortex, hippocampus, substantia nigra, thalamus, and cerebellum) but also in other organs such as the heart and lungs. In the brain, it is widely found in the presynaptic terminals in addition to the cytosol. Under normal conditions, alpha-synuclein is thought to be involved in neurotransmitter release and has been shown to have several neuroprotective functions including suppression of apoptosis, regulation of glucose and dopamine, and antioxidant functions among others75. However, under pathologic conditions, alpha-synuclein misfolds forming fibrils that deposit as Lewy bodies. It is unclear why alpha-synuclein aggregates; as with other forms of dementia, oxidative stress and mutations are thought to play a role76. Lewy pathology is accompanied by the substantial neuronal loss that has been documented in the substantia nigra, locus coeruleus and basal nucleus of Meynert, among other regions77.
In addition to alpha-synuclein, Aβ plaques and tau tangles are often present in LBD78. Similar to the discussion above regarding Aβ and tau, questions about whether alpha-synuclein is neuroprotective or neurotoxic have arisen. In support of a neuroprotective role, Lewy bodies have been shown to isolate harmful proteins79,80 (which could include persistent pathogens) similar to the antimicrobial role of Aβ39,57,58. In addition, inclusion body formation does not necessarily lead to cell death79. Further, there is evidence of severe alpha-synuclein pathology in the absence of clinical symptoms81, highlighting a disconnect between alpha-synuclein pathology and dementia.
LBD Risk Factors
A number of genetic risk factors have been associated with LBP dementia including mutations in SNCA, the gene that encodes the alpha-synuclein protein, as well as genes implicated in other forms of dementia such as those associated with amyloid precursor protein, presenilin genes, tau, and apoE74.
LBD Imaging
Like AD, LBD is associated with global atrophy, rendering structural imaging less helpful in differentiating LBD from AD. Other neuroimaging techniques, however, have proven more useful in terms of differential diagnosis of LBD from AD74. Briefly, LBD, in particular, has been associated with occipital hypometabolism, less hippocampal atrophy, and significantly reduced dopamine reuptake in the caudate and putamen relative to AD.
LBD Treatment
Like AD, treatment of LBD is aimed at symptom management, and acetylcholinesterase inhibitors are commonly prescribed to slow the progression of cognitive deficits. As noted in a recent review, treatments targeting other symptoms have insufficient evidence regarding efficacy and, in many cases, evidence of adverse effects limit the benefit of several medications aimed at symptom management74. As with other proteinopathies, treatments aimed at decreasing the accumulation of alpha-synuclein are being investigated as the future of LBD treatment82.
Vascular Dementia
Vascular dementia (VaD), sometimes referred to as vascular cognitive impairment, accounts for about 15% of dementia cases83. VaD results from reduced blood flow to the brain resulting in cortical and/or subcortical damage. Although the symptoms of VaD are variable depending on which area of the brain is affected by vascular pathology, deficits in planning and judgment are commonly observed (for an overview of VaD, see 84). Depression is also particularly common in VaD. Unlike AD, where memory impairment is a hallmark, the memory may or may not be impaired in VaD. The symptom course is also variable. Some experience sudden onset of symptoms, particularly following a stroke, some exhibit a stepwise progression of symptoms and others a slow steady progression similar to that seen in AD. Finally, VaD and AD often co-occur with up to 50% of individuals showing evidence of “mixed dementia”85, making it among the most common in older adults.
VaD Risk Factors
Several vascular dementia risk factors mirror those associated with heart disease including hypertension, high cholesterol, diabetes, smoking, sedentary lifestyle, unhealthy diet, being overweight, limited exercise, and high alcohol consumption. Notably, these factors have been associated with inflammation which has been shown to contribute to atherosclerosis and subsequent risk of stroke and cognitive impairment86,87. Stroke history is a significant risk factor for vascular dementia with nearly 25% of people who have had a stroke ultimately developing dementia88. As with other forms of dementia, increasing age is a risk factor for vascular dementia. Identification of genetic risk factors for VaD has been limited84; however, like AD, apoE4 has been associated with VaD89.
VaD Neuroimaging and Neuropathology
The presence of infarcts, lacunes, and white matter lesions as evidenced by neuroimaging are indicative of VaD88,90. However, due to the heterogeneity of cerebrovascular pathology and evidence of vascular brain pathology in non-demented older individuals91, there are no accepted neuropathological criteria specific for VaD.
VaD Treatment
To date, pharmacological interventions for VaD have shown limited effectiveness84. Interventions for AD, such as cholinesterase inhibitors and memantine, are not recommended due to side effects and small magnitude benefits observed in VaD relative to AD. Other pharmacological interventions aimed at reducing risk factors such as high cholesterol or hypertension have typically not included cognitive status as a primary outcome, rendering conclusions about effectiveness in reducing dementia risk untenable. Preventive lifestyle interventions such as healthy eating and increased exercise have shown promising results in terms of improving or maintaining cognitive functioning in at-risk elderly people92. The effectiveness of lifestyle interventions is at least partially attributable to reductions in peripheral inflammation93.
Frontotemporal Dementia
Frontotemporal dementia (FTD) is the third most common cause of dementia, with prevalence ranging from 3-26% of dementia cases, and is particularly common in early-onset dementia94. Three variants of FTD have been identified based on early symptoms and are reviewed elsewhere95. First, behavioral–variant FTD is characterized by personality changes, disinhibition, and apathy, often resembling psychiatric disorders. Unlike AD where memory impairment is prominent, memory and visuospatial skills are often spared in the early stages of behavioral-variant FTD. There may be a slow progression of cognitive impairment and neuroimaging is often unremarkable. Primary progressive aphasias are characterized primarily by a decline in language skills during the initial phase of the disease. In semantic-variant primary progressive aphasia, symptoms such as to include anomia, word-finding difficulties, and impaired comprehension. Non-fluent variant primary progressive aphasia is characterized by grammatical errors and speech apraxia. As any of the variants of FTD progress, the symptoms converge and cognitive and motor deficits emerge, ultimately resembling AD.
FTD Risk Factors
A family history of dementia is associated with FTD. To that end, a number of genetic mutations including, but not limited to, mutations associated with tau, TAR DNA-binding protein-43 (TDP-43), fused-in sarcoma (FUS), and ubiquitin have been associated with FTD. A succinct review of several genetic mutations associated with FTD, brain pathology that is characteristic of each mutation, and mechanisms of degeneration is available in a recent FTD overview95. Notably, several studies have noted strong associations between HLA Class II DR and FTD including evidence of cis-changes in methylation levels of HLA-DRA in the frontal cortex96 and a significant reduction in HLA-DR expression if FTD relative to controls97.
FTD Neuropathology
Similar to AD, abnormal protein deposition has been implicated in FTD. Tau, TAR DNA-binding protein-43 (TDP-43), or FUS protein account for most cases of FTD and are associated with characteristic neuropathological signs (e.g, Pick bodies, tufted astrocytes, as described elsewhere95.
FTD Imaging
Neuroimaging studies have demonstrated similarities across FTD variants as well as unique features. Atrophy in ventromedial and posterior orbital frontal regions, insula, and anterior cingulate cortex have been demonstrated across FTD variants; however, the behavioral variant is predominantly associated with dorsolateral frontal atrophy whereas the semantic variant is primarily associated with bilateral anterior temporal atrophy98. The selective frontal and temporal lobe neurodegeneration in FTD has been attributed to the degeneration of von Economo neurons and fork cells which are thought to integrate cortical and subcortical networks; degeneration of von Economo neurons and fork cells are not found in AD or healthy brains99. In addition, as summarized elsewhere95, amyloid tracer imaging, diffusion tensor imaging, and functional connectivity studies have proven useful in distinguishing FTD from AD.
FTD treatment
Treatment for FTD is often aimed at reducing behavioral symptoms with common psychiatric medications, particularly selective serotonin reuptake inhibitors, widely prescribed. Novel treatments aimed at inhibiting aggregation of tau and other proteins are under investigation.
Mixed Dementia
Further complicating research aimed at distinguishing types of dementia is the fact that dementias often co-occur100-102. VaD and AD, in particular, have been shown to co-occur with high rates of overlapping brain pathology (i.e., both evidence of vascular damage along with amyloid plaques and neurofibrillary tangles), sometimes in addition to evidence of Lewy bodies102. The high rates of overlap point to a combination of different neurodegenerative processes that underlie cognitive deterioration associated with dementia.
Prion Dementia
Prion diseases such as Creutzfeldt-Jakob disease (CJD) represent a rare type of dementia caused by misfolded, self-propagating, rapidly accumulating prion proteins, the result of which is neurodegeneration103,104. In contrast to most other dementias that progress slowly, prion dementias are often characterized by rapid progression with death occurring within a few months to years of onset. Prion diseases are most often spontaneous (80-95%) but can also be genetic (familial; 10-15%) or, on rare occasions, acquired through accidental transmission (e.g., surgical or ingestion of infected meat; 1%)105. Typical symptoms of spontaneous CJD, the most common prion disease, include imbalance and incoordination, memory loss and impaired thinking, and psychiatric symptoms such as anxiety or depression; however, CJD affects many areas of the brain and its presentation is variable and often initially difficult to distinguish from neurologic or psychiatric conditions.
Prion Dementia Risk Factors
Mutations in the human prion protein gene, PRNP, that make the protein more susceptible to misfolding, appear to be associated with various forms of prion diseases104.
Prion Dementia Neuropathology
Neuropathological findings of prion diseases include varying severity of atrophy, vacuolation, astrocytic gliosis, and prion protein amyloid plaque deposition. Prion-related neuropathological findings are reviewed elsewhere104,105. Notably, prions tend to co-localize with amyloid plaques106 and have been shown to promote plaque formation107.
Prion Dementia Imaging
Brain MRI has been shown to have high diagnostic utility for sporadic CJD105. Specifically, restricted diffusion in cortical or deep nuclei gray matter and hyperintensity in cortical gyri, caudate, putamen, or thalamus have been associated with both sporadic and familial forms of CJD; however, gross abnormalities may also be absent. Acquired CJD can be distinguished from other human prion diseases by the “pulvinar sign,” in which the pulvinar is brighter than the anterior putamen.
Prion Dementia Treatment
There are no treatments for prion diseases and the often rapid progression of the disease renders clinical trials a significant challenge. Similar to other neurodegenerative disorders, however, pharmacological interventions are currently being investigated along several fronts including immunotherapy and prevention of prion protein aggregation with doxycycline, thus far with mixed or negative results108. Promising initial studies aimed at preventing prion protein conformational changes have shown evidence of decreased vacuolation and prion protein deposition along with slowed neurological and psychiatric symptom progression in mice and macaques109. Effective treatments in humans remain to be discovered.
Conclusions
Growing evidence suggests pathogens may play a crucial role in the development of dementia. Compelling evidence over the last several decades has strongly implicated pathogens in AD and emerging evidence suggests that pathogens may be involved in other dementias as well. We have hypothesized that exposure to pathogens in the absence of immunogenetic protection against those pathogens (due to HLA-antigen) incongruence leads to dementia20. Conversely, an HLA-antigen match with sufficient binding affinity and ability to mount an immune response may protect against dementia18,20. This perspective focuses on the evolutionary protective role HLA plays in the elimination of foreign pathogens to facilitate host protection against a variety of conditions including dementia.
Acknowledgments
Support was provided by the University of Minnesota (the Kunin Professorship for Women's Healthy Brain Aging, the Brain and Genomics Fund, the McKnight Presidential Chair of Cognitive Neuroscience, and the American Legion Brain Sciences Chair) and the U.S. Department of Veterans Affairs. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
References
- World Health Organization. (2017). Dementia: Fact sheet. Retrieved from http://www.who.int/mediacentre/factsheets/fs362/en/
- Chen K, Reiman EM, Alexander GE, et al. Correlations between apolipoprotein E4 gene dose and whole brain atrophy rates. Am J Psychiatry. 2007; 164: 916-921.
- Canuet L, Tellado I, Couceiro V, et al. Resting-state network disruption and APOE genotype in Alzheimer's disease: a lagged functional connectivity study. PLoS One. 2012; 7(9): e46289.
- Leuthold AC, Mahan MY, Stanwyck JJ, et al. The number of cysteine residues per mole in apolipoprotein E affects systematically synchronous neural interactions in women's healthy brains. Exp Brain Res. 2013; 226: 525-536.
- Corder EJ, Saunders AM, Strittmetter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993; 261: 921-923.
- Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013; 45: 1452-1458.
- Wisdom NM, Callahan JL, Hawkins KA. The effects of apolipoprotein E on non-impaired cognitive functioning: a meta-analysis. Neurobiol Aging. 2011; 32: 63-74.
- Liu CC, Kanekiyo T, Xu H, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013; 9: 106-118.
- Bettencourt A, Carvalho C, Leal B, et al., The Protective Role of HLA-DRB113 in Autoimmune Diseases. J Immunol Res. 2015; e948723.
- Furukawa H, Oka S, Tsuchiya N, et al. The role of common protective alleles HLA-DRB1* 13 among systemic autoimmune diseases. Genes and Immunity. 2017; 18: 1-7.
- Georgopoulos AP, James LM, Mahan MY, et al. Reduced Human Leukocyte Antigen (HLA) protection in Gulf War Illness (GWI). EBioMedicine. 2016; 3: 79-85.
- Georgopoulos AP, James LM, Carpenter AF, et al. Gulf War illness (GWI) as a neuroimmune disease. Exp Brain Res. 2017; 235(10): 3217-3225.
- James LM, Christova P, Engdahl BE, et al. Human leukocyte antigen (HLA) and Gulf War Illness (GWI): HLA-DRB1*13:02 spares subcortical atrophy in Gulf War veterans. EBioMedicine. 2017; 26: 126-131.
- Christova P, James LM, Engdahl BE, et al. Subcortical brain atrophy in Gulf War Illness. Exp Brain Res. 2017; 235: 2777-2786.
- Fjell AM, Walhovd KB, Fennema-Notestine C, et al. One-year brain atrophy evident in healthy aging. J Neurosci. 2009; 29(48):15223-15331.
- James LM, Christova P, Lewis SM, et al. Protective effect of Human Leukocyte Antigen (HLA) Allele DRB1* 13: 02 on age-related brain gray matter volume reduction in healthy women. EBioMedicine. 2018 Mar 1; 29: 31-7.
- James LM, Dolan S, Leuthold AC, et al. The effects of human leukocyte antigen DRB1* 13 and apolipoprotein E on age-related variability of synchronous neural interactions in healthy women. EBioMedicine. 2018; 35: 288-94.
- James LM, Georgopoulos AP. The human leukocyte antigen (HLA) DRB1*13:02 allele protects against dementia in Continental Western Europe. J Neurol Neurosci. 2019; 4(5): 1-6.
- James LM, Georgopoulos AP. Persistent Antigens Hypothesis: The Human Leukocyte Antigen (HLA) Connection. J Neruol Neuromed. 2018; 3(6): 27-31.
- James LM, Georgopoulos AP. Human leukocyte antigen as a key factor in preventing dementia and associated apolipoprotein E4 risk. Front Aging Neurosci. 2019; 11: 82.
- Mahley RW, Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012; 76(5): 871-85.
- Itzhaki RF. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer's disease. Frontiers in aging neuroscience. 2018; 10: 324.
- Mawanda F, Wallace R. Can infections cause Alzheimer’s disease? Epidemiol Rev. 2013; 35: 161-80.
- Jang H, Boltz D, Sturm-Ramirez K, et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. PNAS. 2009; 106(33): 14063-8.
- Kinney JW, Bemiller SM, Murtishaw AS, et al. Inflammation as a central mechanism in Alzheimer's disease. Alzheimer's & Dementia: Translational Research & Clinical Interventions. 2018; 4: 575-90.
- Kuo HK, Yen CJ, Chang CH, et al. Relation of C-reactive protein to stroke, cognitive disorders, and depression in the general population: systematic review and meta-analysis. Lancet Neurol. 2005; 4: 371–80.
- Sjögren M, Folkesson S, Blennow K, et al. Increased intrathecal inflammatory activity in frontotemporal dementia: pathophysiological implications. J Neurol Neurosur Psychiat. 2004; 75(8): 1107-11.
- Raz N, Rodrigue KM. Differential aging of the brain: patterns, cognitive correlates and modifiers Neurosci Biobehav Rev. 2006; 30: 730-748.
- Dando SJ, Mackay-Sim A, Norton R. Pathogens Penetrating the Central Nervous System: Infection Pathways and the Cellular and Molecular Mechanisms of Invasion. Clin Microbiol Rev. 2014; 27(4): 691–726.
- Mori I, Goshima F, Imai Y, et al. Olfactory receptor neurons prevent dissemination of neurovirulent influenza A virus into the brain by undergoing virus-induced apoptosis. J Gen Virol. 2002; 83: 2109–2116.
- Alves J, Petrosyan A, Magalhães R. Olfactory dysfunction in dementia. World Journal of Clinical Cases WJCC. 2014; 2(11): 661.
- Itzhaki RF, Lin WR, Shang D, et al. Herpes simplex virus type 1 in brain and risk of Alzheimer's disease. The Lancet. 1997; 349(9047): 241-4.
- McQuillan G, Kruszon-Moran D, Flagg EW, et al. Prevalence of herpes simplex virus type 1 and type 2 in persons aged 14–49: United States, 2015–2016. NCHS Data Brief, no 304. Hyattsville, MD: National Center for Health Statistics. 2018.
- Matthews KA, Xu W, Gaglioti AH, et al. Racial and ethnic estimates of Alzheimer's disease and related dementias in the United States (2015–2060) in adults aged ≥65 years. Alzheimers Dement. 2019; 15: 17– 24.
- Wozniak MA, Itzhaki RF, Shipley SJ, et al. Herpes simplex virus infection causes cellular β-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007; 429: 95–100.
- Wozniak MA, Frost AL, Itzhaki RF. Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimers Dis. 2009; 16: 341–350.
- Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol. 2009; 217: 131–138.
- Canet G, Dias C, Gabelle A, et al. HIV neuroinfection and Alzheimer's disease: Similarities and potential links? Front Cell Neurosci. 2018; 12: 307.
- Li H, Liu CC, Zheng H, et al. Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer's disease -conformist, nonconformist, and realistic prospects for AD pathogenesis. Transl Neurodegeneration. 2018; 7: 34.
- Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018; 14(12): 1602–14.
- Castellani RJ, Lee HG, Perry G, et al. Antioxidant protection and neurodegenerative disease: The role of amyloid-β and tau. Am J Alzheimers Dis. 2006; 21(2): 126-130.
- Soares BP, Provenzale JM. Imaging of herpesvirus infections of the CNS. AJR Am J Roentgenol. 2016; 206: 39–48.
- Milanini B, Valcour V. Differentiating HIV-associated neurocognitive disorders from Alzheimer’s disease: an emerging issue in geriatric NeuroHIV. Current HIV/AIDS Reports. 2017; 14(4): 123-32.
- Chen VCH, Wu SI, Huang KY, et al. Herpes zoster and dementia. J Clin Psychiat. 2018; 79(1): e16m11312.
- Tzeng NS, Chung CH, Lin FH, et al. Antiherpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in Taiwan. Neurotherapeutics. 2018; 15: 417–429.
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2019; 15(3): 321-87.
- Carmona S, Hardy J, Guerreiro R. The genetic landscape of Alzheimer disease. In Handbook of Clinical Neurology. 2018; Vol. 148: 395-408. Elsevier 2018.
- Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention intervention and care. The Lancet. 2017; 390(10113): 2673-734.
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Bio. 2007; 8(2): 101-112.
- Aoyagi A, Condello C, Stöhr J, et al. Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci Transl Med. 2019; 11: eaat8462.
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010; 11(4): 301.
- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013; 501: 45–51.
- Ricciarelli R, Fedele E. The amyloid cascade hypothesis in Alzheimer's Disease: It's time to change our mind. Curr Neuropharmacol. 2017; 15(6): 926–935.
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999; 45(3): 358-68.
- Wolf DS, Gearing M, Snowdon DA, et al. Progression of regional neuropathology in Alzheimer disease and normal elderly: findings from the Nun study. Alz Disease Assoc Dis. 1999; 13(4): 226-31.
- Aizenstein HJ, Nebes RD, Saxton JA, et al., Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008; 65: 1509-1517.
- Bourgade K, Dupuis G, Frost EH, et al. Anti-viral properties of amyloid- β peptides. J Alzheimers Dis. 2016; 54: 859–878.
- Kumar DK, Choi SH, Washicosky KJ, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016; 8: 340ra72.
- Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. PNAS. 1986; 83: 4913–4917.
- Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. 2018; 69: 437–49.
- Rodriguez-Rodriguez P, Sandebring-Matton A, Merino-Serrais P, et al. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain. 2017; 140: 3269–85.
- Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003; 60: 1495–500.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991; 82: 239–59.
- Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011; 70: 960-969.
- Jack CR Jr, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol. 2017; 16: 435–44.
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999; 58: 188–97.
- Lee HG, Perry G, Moreira PI, et al. Tau phosphorylation in Alzheimer's disease: pathogen or protector?. Trends Molec Med. 2005; 11(4): 164–169.
- Pini L, Pievani M, Bocchetta M, et al. Brain atrophy in Alzheimer’s disease and aging. Ageing Res Rev. 2016; 30: 25-48.
- Poulakis K, Pereira JB, Mecocci P, et al. Heterogeneous patterns of brain atrophy in Alzheimer's disease. Neurobiol Aging. 2018; 65: 98-108.
- Murray IVJ, Lee VMY, Trojanowski JQ. Synucleinopathies: A pathological and molecular review. Clin Neurosci Res. 2001; 1: 445.
- Femminella G, Thayanandan T, Calsolaro V, et al. Imaging and Molecular Mechanisms of Alzheimer’s Disease: A Review. Int J Mol Sci. 2018; 19(12): 3702.
- Ballard C, Gauthier S, Corbett A, et al. Alzheimer's disease. The Lancet. 2011; 377(9770): 1019-31.
- Savica R, Grossardt BR, Bower JH, et al. Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol. 2013; 70: 1396.
- Walker Z, Possin KL, Boeve BF, et al. Lewy body dementias. The Lancet. 2015; 386(10004): 1683-97.
- Emamzadeh FN. Alpha-synuclein structure, functions, and interactions. J Res Med Sci. 2016; 21: 29.
- Benskey MJ, Perez RG, Manfredsson FP. The contribution of alpha synuclein to neuronal survival and function—implications for Parkinson’s disease. J Neurochem. 2016; 137: 331–359.
- Zarrow C, Lyness SA, Mortimer JA, et al. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003; 60: 337–41.
- Irwin DJ, Lee VM, Trojanowski JQ. Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci. 2013; 14(9): 626-36.
- Tanaka M, Kim YM, Lee G, et al. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004; 279: 4625–4631.
- Castellani RJ, Siedlak SL, Perry G, et al. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta neuropathologica. 2000; 100(2): 111-4.
- Parkkinen L, Pirttila T, Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008; 115: 399–407.
- Masliah E, Rockenstein E, Mante M, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011; 6: e19338.
- Ott A, Breteler MM, van Harskamp F, et al. Prevalence of Alzheimer’s disease and vascular dementia: association with education The Rotterdam study. BMJ. 1995; 310: 970–973.
- T O'Brien J, Thomas A. Vascular dementia. The Lancet. 2015; 386(10004): 1698-706.
- Schneider JA, Arvanitakis Z, Bang W, et al. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007; 69(24): 2197-204.
- Napoli C, Palinski W. Neurodegenerative diseases: insights in to pathogenetic mechanisms from atherosclerosis. Neurobiol Aging. 2005; 26(3): 293-302.
- Ravaglia G, Forti P, Maioli F, et al. Blood inflammatory markers and risk of dementia: The Conselice Study of Brain Aging. Neurobiol Aging. 2007; 28(12): 1810-20.
- Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009; 8: 1006–18.
- Dwyer R, Skrobot OA, Dwyer J, et al. Using Alzgene-like approaches to investigate susceptibility genes for vascular cognitive impairment. J Alzheimers Dis. 2013; 34: 145–54.
- Pohjasvaara T, Mäntylä R, Salonen O, et al. How complex interactions of ischemic brain infarcts, white matter lesions, and atrophy relate to poststroke dementia. Arch Neurol. 2000; 57(9): 1295-300.
- Stephan B, Matthews FE, Ma B, et al. Alzheimer and vascular neuropathological changes associated with different cognitive States in a non-demented sample. J Alz Dis. 2012; 29(2): 309-18.
- Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. The Lancet. 2015; 385(9984): 2255-63.
- Ford E. Does exercise reduce inflammation? Physical activity and C-reactive protein among U.S. adults. Epidemiology. 2002; 13: 561–568.
- Vieira RT, Caixeta L, Machado S, et al. Epidemiology of early-onset dementia: a review of the literature. Clin Prac Epidemiol Ment health. 2013; 9: 88-95.
- Bang J, Spina S, Miller BL. Frontotemporal dementia. The Lancet. 2015; 24386(10004): 1672-82.
- Ferrari R, Hernandez DG, Nalls MA, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014; 13: 686–699.
- Busse M, Michler E, von Hoff F, et al. Alterations in the peripheral immune system in dementia. J Alzheimers Dis. 2017; 58: 1303–1313.
- Rosen HJ, Gorno–Tempini ML, Goldman WP, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002; 58(2): 198-208.
- Kim EJ, Sidhu M, Gaus SE, et al. Selective frontoinsular von Economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb Cortex. 2012; 22: 251–59.
- Korczyn, AD. Mixed dementia—the most common cause of dementia. Ann NY Acad Sci. 2002; 977(1): 129-134.
- Langa KM, Foster NL, Larson EB. Mixed dementia: emerging concepts and therapeutic implications. JAMA. 2004; 292(23): 2901-2908.
- Schneider JA. High blood pressure and microinfarcts: a link between vascular risk factors, dementia, and clinical Alzheimer's disease. J Am Geriatr Soc. 2009; 57(11): 2146-7.
- Prusiner SB. Prions. PNAS. 1998; 95(23): 13363–13383.
- Prusiner SB. Neurodegenerative diseases and prions. N Engl J Med 2001; 344: 1516-1526.
- Geschwind MD. Prion diseases. Continuum Minneap. Minn. 2015; 21:1612–1638.
- Voigtlander T, Kloppel S, Birner P, et al. Marked increase of neuronal prion protein immunoreactivity in Alzheimer's disease and human prion diseases. Acta Neuropathol. 2001; 101: 417–423.
- Schwarze-Eicker K, Keyvani K, Gortz N, et al. Prion protein (PrPc) promotes beta-amyloid plaque formation. Neurobiol Aging. 2005; 26: 1177–1182.
- Forloni G, Salmona M, Marcon G, et al. Tetracyclines and prion infectivity. Infectious Disord Drug Targets. 2009; 9(1): 23-30.
- Yamaguchi K, Kamatari YO, Ono F, et al. A designer molecular chaperone against transmissible spongiform encephalopathy slows disease progression in mice and macaques. Nat Biomed Engineer. 2019; 3(3): 206.