
When transporters fail to be transported:how to rescue folding-deficient SLC6 transporters
Sonja Sucic, Ameya Kasture, H. M. Mazhar Asjad, Carina Kern, Ali El-Kasaby and Michael Freissmuth*
Institute of Pharmacology, Center of Physiology and Pharmacology, Medical University of Vienna, A-1090 Vienna, Austria
Abstract
The human dopamine transporter (hDAT) belongs to the solute carrier 6 (SLC6) gene family. Point mutations in hDAT (SLC6A3) have been linked to a syndrome of dopamine transporter deficiency or infantile dystonia/parkinsonism. The mutations impair DAT folding, causing retention of variant DATs in the endoplasmic reticulum and subsequently impair transport activity. The folding trajectory of DAT itself is not understood, though many insights have been gained from studies of folding-deficient mutants of the closely related serotonin transporter (SERT); i.e. their functional rescue by pharmacochaperoning with (nor)ibogaine or heat-shock protein inhibitors. We recently provided a proof-of-principle that folding-deficits in DAT are amenable to rescue in vitro and in vivo. As a model we used the Drosophila melanogaster DAT mutant dDAT-G108Q, which phenocopies the fumin/sleepless DAT-knockout. Treatment with noribogaine and/or HSP70 inhibitor pifithrin-μ restored folding of, and dopamine transport by, dDAT-G108Q, its axonal delivery and normal sleep time in mutant flies. The possibility of functional rescue of misfolded DATs in living flies by pharmacochaperoning grants new therapeutic prospects in the remedy of folding diseases, not only in hDAT, but also in other SLC6 transporters, in particular mutants of the creatine transporter-1, which give rise to X-linked mental retardation.
Point mutations in a gene, which result in misfolding of the encoded protein, are known to be the underlying cause of many diseases. In fact, the term molecular medicine was coined in conjunction with the study of sickle cell anaemia, the prototypical protein folding disease1. With a few prevalent exceptions (e.g. sickle cell anaemia), individual folding diseases are rare (i.e., affecting less than 1 in 2000 persons), but collectively they affect many people. In addition and more importantly, it has been pointed out more than 75 years ago that the study of rare diseases is intrinsically worthwhile: major scientific advances have been achieved by studying rare diseases2. We posit that this also true for the study of misfolded versions of neurotransmitter transporters of the SL6 family. The human genome encodes twenty members of the solute carrier 6 (SLC6) gene family, but SLC6A10 (creatine transporter-2/CT-2) is a pseudogene: SLC6 transporters can be grouped into 4 families based on their evolutionary relation, namely (i) the monoamine transporters for dopamine (DAT, SLC6A3), serotonin (SERT, SLC6A4) and noradrenaline (NET, SLC6A2), (ii) the amino acid/neurotransmitter transporters (e.g. glycine tranporters-1 and -2 = SLC6A9 and SLC6A5, respectively), the GABA/osmolyte-transporters (including GABA-transporters GAT-1 to -4 = SLC6A1, SLC6A13, SLCA11 & SLCA12) and (iv) the nutrient/orphan amino acid transporters3.
SLC6 transporters operate as NaCl-dependent transporters; they exploit the electrochemical gradient for Na+ to drive inward transport of substrate in a conformational cycle comprising an outward-facing conformation, a substrate- and Na+- and Cl--bound (stoichiometry 2 or 3 Na+, 1 Cl-) occluded state, an inward-facing conformation and a K+- and Cl--bound return step4. In addition, SLC6 transporters can also operate in the substrate-exchange mode, which is the basis for amphetamine-induced reverse transport by DAT, NET and SERT; this accounts for the psychostimulant actions of amphetamines5.
Missense mutations, which give rise to a human disease, occur in many SLC6 transporters6. In three instances, these mutations have been shown to cause misfolding of the mutated SLC6 transporter, i.e. in NET, DAT and GlyT26. A single mutation in NET (A457P) causes postural hypotension/orthostatic intolerance, which is genetically transmitted in a dominant fashion7. There are more than 15 mutations in DAT, which result in a folding defect and cause a recessive form of infantile/juvenile dystonia/Parkinson's disease8-10 and at least 8 mutations, which result in misfolding of GlyT2 and thus lead to hyperekplexia/startle disease11-14. Two of these mutations act in dominant manner (see below). Finally, there is one SLC6 transporter, i.e. the creatine transporter-1 (CT-1/SLC6A8), where the circumstantial evidence suggests that - at least in some of the 22 missense mutations15 - the disorder (mental retardation) is also due to misfolding of the mutated transporter (see below). SLC6 transporters epitomise the folding problem of dynamic polytopic membrane proteins: SLC6 transporters have 12 transmembrane spanning (mostly α-helical) segments; thus the bulk of the protein is hydrophobic in nature. If all known mutations of SLC6 transporters, which are known or suspected to cause misfolding, are mapped onto a structural model of SLC6 transporters (based on the available crystal structures of DAT), they are found to be enriched at the lipid/protein-interface6. This indicates that the lipid bilayer imposes a major constraint as the nascent protein moves through the conformational search space to reach a stable fold. This can be rationalized, if the individual steps of the folding trajectory are recapitulated (for details see ref. 6): (i) the transmembrane helices are co-translationally inserted into the SEC61 translocon of the endoplasmic reticulum. At this stage, the motion of amino acid side chains is restricted, thus limiting the search space. (ii) Transmembrane segments are released individually or in pairs of two via a lateral gate of SEC61 into the lipid bilayer16 (Fig. 1a). (iii) Within the lipid bilayer, the transmembrane segments of all polytopic membrane proteins have to rearrange, because they typically adopt an annular rather than a serpentine topology. In nascent SLC6 transporters, this requires lipids to be displaced from those surfaces of the 12 α-helical transmembrane segments, which face each other or from the translocation pathway (Fig. 1b). The annular arrangement must be bolted to reach the stable fold. (iv) Finally, it is worth considering that SLC6 transporters have - by definition - a flexible conformation, because they must support the transport cycle (see above). The question thus arises whether the folding trajectory proceeds to the outward or the inward facing conformation.
A keyhole perspective of the folding problem:
It is obvious that the folding trajectory of SLC6 transporters must move through a conformational search space but the underlying details are not known. However, several serendipitous insights offer a glimpse of the problem and allow for generating testable models. In addition, the SLC6 folding mutants, which cause human diseases, provide a backdrop to examine the models for their explanatory power. The first relevant finding is the observation that SLC6 transporters form constitutive oligomers17. The oligomers are kinetically trapped at the cell surface, i.e. they do not exchange18 but not within the endoplasmic reticulum (ER)19. Thus, oligomerization occurs in the ER. Oligomerization in the ER is a prerequisite for ER-export20; conversely, export-deficient versions of GAT1 or of SERT exert a dominant-negative effect on the wild type protein21,22. Thus, based on these findings it is possible to rationalize the dominant negative effect of misfolded SL6 transporters such as NET-A457P7, GlyT2-S510R and related mutants11,13,14.
However, this does not explain the recessive transmission of the other hyperekplexia-causing mutations in GlyT211,12 or all known mutations in DAT, which give rise to childhood dystonia/parkinsonism8-10.The apparent oxymoron of having both, dominant and recessive SLC6 mutations, can be resolved by considering that oligomer formation occurs late in the folding trajectory. In fact, the available evidence supports a model, where the nascent SLC6 transporter is engaged by several chaperones in the ER lumen, most notably calnexin23 and a cytosolic chaperone relay, which engages the C-terminus of the transporter24 (Fig. 1b and Fig. 1c). Based on this model6, it is possible to rationalize how a recessive transmission operates: the misfolded SLC6 transporter is trapped by ER-resident lumenal chaperones (e.g. calnexin), which precludes oligomer formation with the product of the wild type allele.
One way to approach the folding problem is to work backwards from the folded state and ask, which conformation(s) was/were visited before the folded state was reached. A serendipitous finding was the observation that the drug noribogaine corrected the folding deficit of several SERT mutants, which had been created to study ER export25 and the folding problem24. Ibogaine and its derivative noribogaine bind to and stabilize the inward facing conformation26,27. This implies that the folding trajectory proceeds through the inward facing conformation. Accordingly, mutations, which trap the transporter in the inward facing state, are predicted to remedy the folding deficit. Several inward-facing mutant versions of SERT are available28,29. If these mutations are introduced into folding-deficient mutants of SERT, they act as second site suppressors, i.e. they restore folding and promote ER export of the double mutants, although the extent to which individual mutants are rescued differs30. Thus, the available evidence suggests that the folding trajectory moves through the inward facing conformation.
As mentioned above, the annular arrangement of the transmembrane helices must be stabilized during the folding trajectory. It was appreciated more than a decade ago that serial truncation of GAT131 or of SERT32 impaired surface expression. Similarly, mutations within the first intracellular loop of NET also affect the delivery of the transporter to the plasma membrane33-35. These two sets of information can be rationalized: both, the first intracellular loop and the C-terminus are required for folding, because a salt bridge is formed between the end of a helical segment in the C-terminus and the first intracellular loop; this interaction presumably stabilizes the annular arrangement of the hydrophobic core and thus facilitates folding of SERT30. The C-terminus of SERT is shielded by a heat-shock protein relay24: folding-deficient mutants are stalled in different complexes. This association provides a handle to assess progression of SERT and - by inference - of other SLC6 transporters through the folding trajectory24,30. In addition and importantly, this insight allows for targeting the folding machinery with drugs (Fig. 1; see also below).
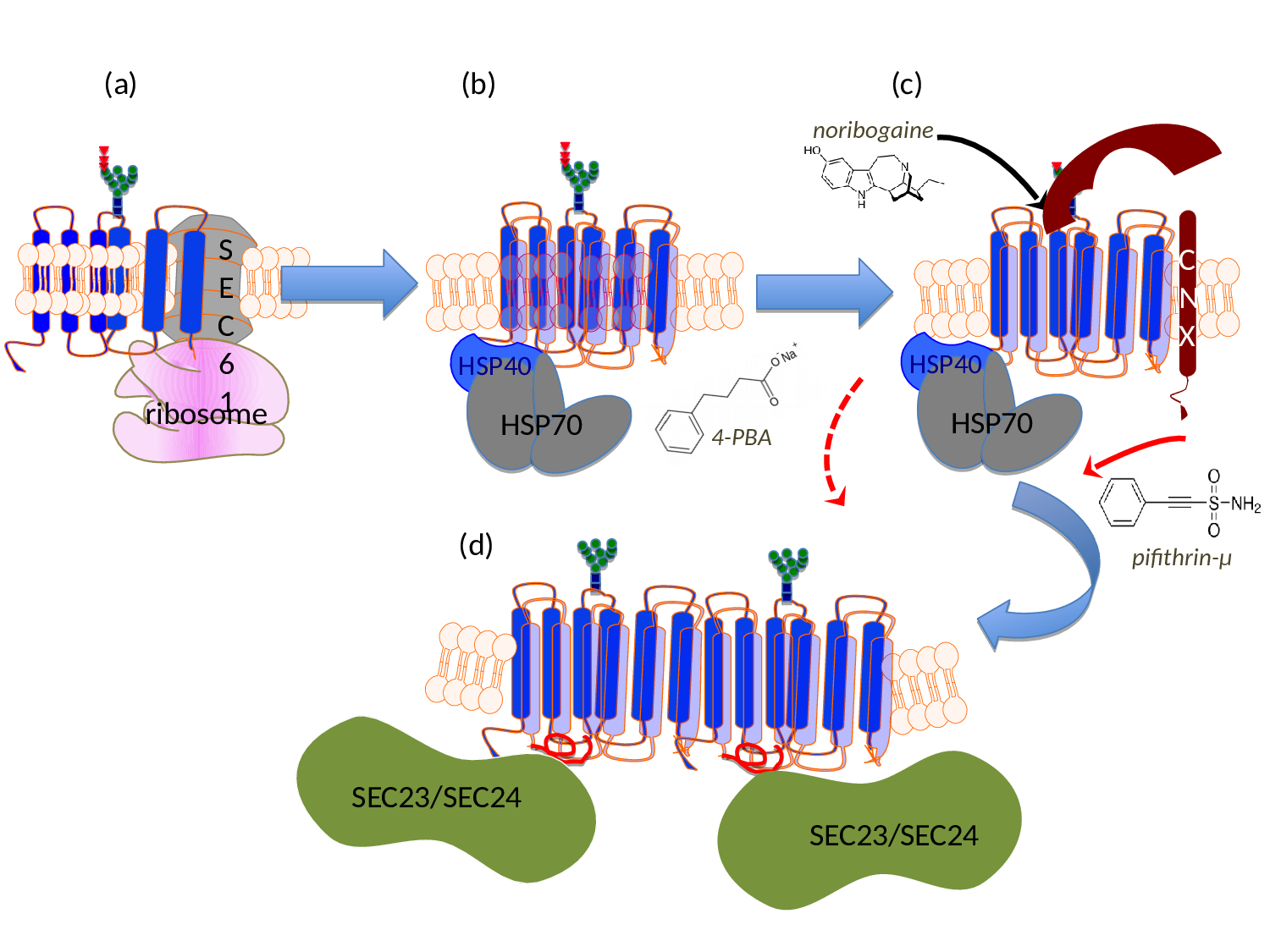
Figure 1:Schematic representation of the sites of action of pharmacochaperones and chemical chaperones in the folding trajectory of DAT. (a) The first transmembrane segment of nascent DAT acts as a signal peptide. The ribosome is recruited to the membrane of the endoplasmic reticulum (ER) via the signal recognition particle (SRP) and the SRP receptor (not shown), where the translation arrest is lifted and the transmembrane helices are cotranslationally inserted into the translocon/SEC61 channel. The helices are released into the ER membrane via a lateral gate. On the lumenal side (topologically equivalent to the extracellular side), the nascent protein chain is subject to N-linked core glycosylation (blue squares represent N-actely-glucosamin, green dots mannose and triangles glucose). (b) To achieve an annular arrangement, lipids have to be expelled from the interior of the ring. On the cytosolic side, the C-terminus is engaged by a heat-shock protein relay (shown here is a dimer of HSP40 and HSP70) to promote folding and to preclude premature engagement of the COPII-coat. (c) During folding, ER-resident, lumenal chaperones are recruited to folding intermediates: shown here is calnexin (CNX), which recognizes the (re)glucosylated folding intermediates via its lectin domain. (d) When the minimum energy conformation - i.e. the stably folded state - is reached the chaperones are released: the transporter forms an oligomer and the cognate SEC23/SEC24-dimer (containing SEC24D for DAT, see ref. 39) is recruited to the C-terminus, which contains an α-helix (highlighted in red). This C-terminal α-helix interacts with the first intracellular loop and thus bolts the annular arrangement of hydrophobic core. The bow-tie shape of the COPII-component SEC23/SEC24 stabilizes the membrane curvature of the nascent vesicle, which will carry the transporter en route to the Golgi. Noribogaine binds to the ligand/substrate binding-site within the hydrophobic core and stabilizes the inward facing conformation. This lowers the energy barrier between folding intermediates and thus facilitates the progression along the folding trajectory. Pifithrine-µ inhibits HSP70 and is thought to thereby release stalled transporter complexes. 4-Phenylbutyric acid (4-PBA) modulates the expression levels of various HSP70 family members. This is thought to shift the balance in favor of progression through the folding trajectory, while the formation of stalled complexes is reduced.
The acid test - rescuing mutated transporters:
The vast majority of missense mutations in human DAT, which cause childhood dystonia/parkinsonism, give rise to a folding-deficient transporter, which is retained in the ER8-10. The phenotypic consequence is that of dopamine deficiency rather than a hyperdopaminergic state, because vesicular stores of dopamine are not replenished. The acid test for any models, which summarize the rudimentary understanding of SLC6 transporter folding, is their ability to guide attempts to rescue folding-deficient transporter mutants. We translated the insights, which we had gained by studying ER export of GAT120,21,31,36,37, and SERT23,38-40and the rescue of folding deficient mutants24-25,30, to a DAT mutant, which was serendipitously discovered in Drosophila melanogaster: flies harboring Drosophila DAT-G108Q (dDAT-G108Q) have very much reduced sleep time41. Hence this mutation phenocopies the DAT knock-out in flies (referred to as fumin, i.e. sleepless)42. Glycine 108 is part of a GXXXG-related motif, which stabilizes the interaction between TM3 and TM12 at the intracellular leaflet of the membrane. When substituted by the large glutamine, the mutation interferes with packing of helix TM12 in the folding trajectory of dDAT. This in turn affects the C-terminus, which closely follows TM1243. As outlined above, the C-terminus is positioned to stabilize the annular arrangement of the hydrophobic core. Accordingly, in transfected cells, dDAT-G108Q and its human equivalent hDAT-G140Q were trapped in the ER in complex with calnexin and HSP70-1A, reflecting the stalling of the mutant along the folding trajectory43. The folding defect of dDAT-G108Q was remedied by the pharmacochaperone noribogaine or the HSP70 inhibitor pifithrin-μ:, the mutant reached the cell surface, and transport activity was also recovered. Most importantly, this pharmacochaperone action observed in cell cultures was reproduced in vivo in dDAT-G108Q flies: upon pharmacochaperone treatment the mutant reached the axonal projections (Fig. 2) to a level sufficient to recover sleep43. Axonal targeting is obviously important, because the refilling of vesicular stores of neurotransmitters depends on this eponymous action of DAT: mutants of GAT1 and SERT, which fail to recruit their cognate SEC24-isoform eventually do reach the cell surface, but they are not delivered to the presynaptic specialization36,37,40. Thus, the fact that dDAT-G108Q and hDAT-G140Q reached the axonal territory shows that neither pharmacochaperoning by noribogaine nor inhibition of HSP70 by pifithrin-µ reroute the transporter through an atypical ER export pathway. Based on these findings, it was sound to predict that some of the misfolded mutants of DAT ought to be rescued by pharmacochaperoning with noribogaine, and that childhood dystonia/parkinsonism may be amenable to treatment by pharmacochaperones and/or HSP70 inhibition, which restore folding of the mutated DAT-versions. In fact, some aspects of this prediction have already been verified in transfected cells: cell surface expression of several disease-causing DAT-mutants was restored by pharmacochaperoning with ibogaine and with bupropion44, which is of particular interest, because bupropion is an approved drug.
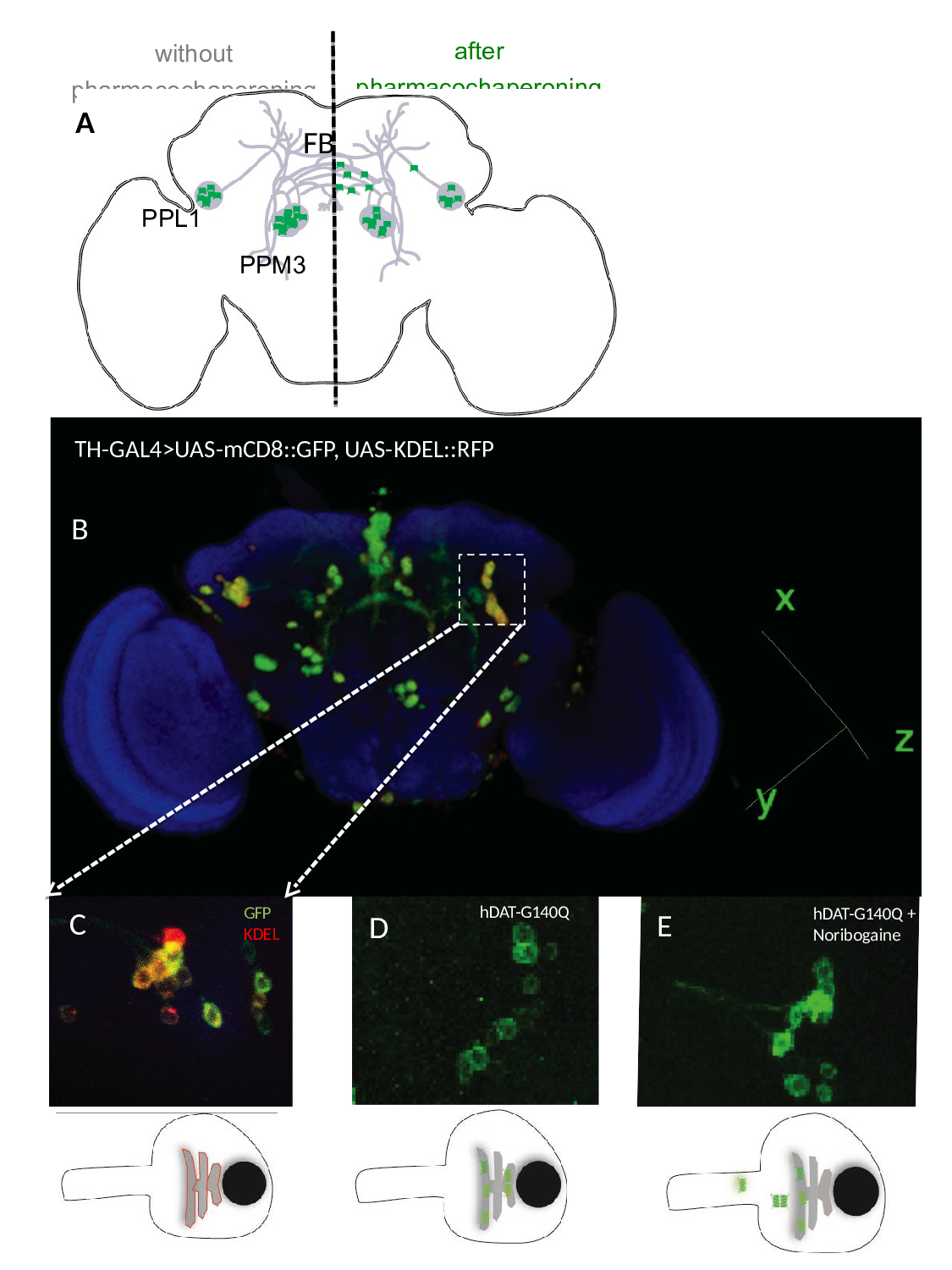
Figure 2:Pharmacochaperoning rescues a misfolded human DAT (hDAT-G104Q) in the brain of Drosophila melanogaster. A) Schematic cartoon showing the dopaminergic dorsomedial posterior protocerebral (PPM3) and dorsolateral posterior protocerebral neurons (PPL1) neurons, which project their axons into the fan-shaped body (FB) of the fly brain. In the absence of pharmacochaperoning (left hand side) the mutant DAT mutant (green dots) is retained in the ER; if flies are administered noribogaine via their food, a considerable fraction of the DAT mutant reaches the presynaptic specialization (right hand side). B) Posterior view of 3D rendered adult fly brain expressing the surface marker mCD8-green fluorescence protein (GFP) and the ER marker red fluorescence protein (RFP)-KDEL under the control of tyrosine hydroxylase GAL4 (TH-GAL4). C) Magnified image of paired posterior lateral 1 (PPL1) cluster of dopaminergic neurons. D and E. TH-GAL4 driven expression of hDAT-G140Q in PPL1 neurons in the brain of untreated flies (D) and flies receiving noribogaine (100 μM) in their food (E). It is evident from panel E that the GFP-tagged (=green) hDAT-G140Q entered the axonal extension, whereas in panel D it is confined to the ER in the cell soma. The bottom panels represent schematic cartoons of the fluorescent images shown above outlining the red fluorescence of KDEL in the ER within the soma (left), the green fluorescence of the DAT mutant in untreated flies (middle), which upon pharmacochaperoning leaves the ER and enters into neurites (right).
The observations on dDAT-G108Q also have repercussions for mutations in creatine transporter-1 (SLC6A8): a mutation of the equivalent glycine (G132V) is found in boys with mental retardation15,45. It is therefore reasonable to assume that CT1-G132V is also misfolded and that is also amenable to rescue by pharmacochaperoning and/or inhibition of heat-shock proteins. The monoamine transporters DAT, SERT and NET have a rich pharmacology5: several hundred inhibitors and substrate analogues are available, which is a treasure trove in the search for pharmacochaperones. In contrast, the number of CT1-ligands is limited. This makes the inhibition of heat-shock proteins or the manipulation of their expression by 4-phenylbutyrate46,47 of particular interest to restore folding and surface expression of mutated versions of CT1.
Acknowledgements
Work from the authors' laboratory is supported by Project Program Grant SFB35-10 (to M. F.) and Grant P27518-B27 (to S. S.) by the Austrian Science Fund/FWF; A.K. and H.H.MA. are supported by the doctoral programme CCHD, which is jointly funded by the FWF and the Medical University of Vienna, and by a PhD-stipend jointly awarded by the Higher Education Commission of Pakistan and the Austrian Agency for International Cooperation in Education and Research/OeAD, respectively.
References
- Pramod AB, Foster J, Carvelli L, et al. SLC6 transporters structure function regulation disease association and therapeutics. Mol Aspects Med. 2013 Apr-Jun; 34(2-3): 197-219.
- Pauling L, Itano HA, Singer SJ, et al. Sickle cell anemia a molecular disease. Science. 1949 Nov 25; 110(2865): 543-548.
- Garrod A. The lessons of rare maladies. Lancet. 1928 May 26;211(5465):1055–1060.
- Hasenhuetl PS, Freissmuth M, Sandtner W. Electrogenic binding of intracellular cations defines a kinetic decision Point in the transport cycle of SERT. J Biol Chem. 2016 Oct 18. pii: jbc.M116.753319.
- Sitte HH, Freissmuth M. Amphetamines new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol Sci. 2015 Jan; 36(1): 41-50.
- Chiba P, Freissmuth M, Stockner T. Defining the blanks pharmacochaperoning of SLC6 transporters and ABC transporters. Pharmacol Res. 2014 May; 83: 63-73.
- Hahn MK, Robertson D, Blakely RD. A mutation in the human norepinephrine transporter gene SLC6A2 associated with orthostatic intolerance disrupts surface expression of mutant and wild type transporters. J Neurosci. 2003 Jun 1; 23(11): 4470–4478.
- Kurian MA, Zhen J, Cheng SY, et al. Homozygous loss of function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism dystonia. J Clin Invest. 2009 Jun; 119(6): 1595-1603.
- Kurian MA, Li Y, Zhen J, et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome an observational cohort and experimental study. Lancet Neurol. 2011 Jan; 10(1): 54-62.
- Ng J, Zhen J, Meyer E, et al. Dopamine transporter deficiency syndrome phenotypic spectrum from infancy to adulthood. Brain. 2014 Apr; 137(Pt 4): 1107-1119.
- Rees MI, Harvey K, Pearce BR, et al.Mutations in the gene encoding GlyT2 SLC6A5 define a presynaptic component of human startle disease. Nat Genet. 2006 Jul; 38(7): 801-806.
- Rees MI, Harvey K, Pearce BR, et al.Mutations in the gene encoding GlyT2 SLC6A5 define a presynaptic component of human startle disease. Nat Genet. 2006 Jul; 38(7): 801-806.
- Giménez C, Pérez Siles G, Martínez Villarreal J, et al. A novel dominant hyperekplexia mutation Y705C alters trafficking and biochemical properties of the presynaptic glycine transporter GlyT2. J Biol Chem. 2012 Aug 17; 287(34): 28986-9002.
- Arribas González E, de Juan Sanz J, Aragón C, et al. Molecular basis of the dominant negative effect of a glycine transporter 2 mutation associated with hyperekplexia. J Biol Chem. 2015 Jan 23; 290(4): 2150-2165.
- van de Kamp JM, Betsalel OT, Mercimek Mahmutoglu S, et al. Phenotype and genotype in 101 males with X linked creatine transporter deficiency. J Med Genet. 2013 Jul; 50(7): 463-472.
- Park E, Rapoport TA. Mechanisms of Sec61/SecY mediated protein translocation across membranes. Annu Rev Biophys. 2012 June; 41: 21– 40.
- Schmid JA, Scholze P, Kudlacek O, et al. Oligomerization of the human serotonin transporter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J Biol Chem. 2001 Feb 9; 276(6): 3805-3810.
- Anderluh A, Klotzsch E, Reismann AW, et al. Single molecule analysis reveals coexistence of stable serotonin transporter monomers and oligomers in the live cell plasma membrane. J Biol Chem. 2014 Feb 14; 289(7): 4387-4394.
- Anderluh A, Klotzsch E, Ries J, et al. Tracking single serotonin transporter molecules at the endoplasmic reticulum and plasma membrane. Biophys J. 2014 May 6; 106(9): L33-L35.
- Korkhov VM, Farhan H, Freissmuth M, et al. Oligomerization of the γ aminobutyric acid transporter 1 is driven by an interplay of polar and hydrophobic interactions in transmembrane helix II. J Biol Chem. 2004 Dec 31; 279(53): 55728-55736.
- Farhan H, Reiterer V, Korkhov VM, et al. Concentrative export from the endoplasmic reticulum of the γ aminobutyric acid transporter 1 requires binding to SEC24D. J Biol Chem. 2007 Mar 9; 282(10): 7679-7689.
- Just H, Sitte HH, Schmid JA, et al. Identification of an additional interaction domain in transmembrane domains 11 and 12 that supports oligomer formation in the human serotonin transporter. J Biol Chem. 2004 Feb 20; 279(8): 6650-6657.
- Korkhov VM, Milan-Lobo L, Zuber B, et al. Peptide-based interactions with calnexin target misassembled membrane proteins into endoplasmic reticulum-derived multilamellar bodies. J Mol Biol. 2008 Apr 25; 378(2): 337-352.
- El Kasaby A, Koban F, Sitte HH, et al. A cytosolic relay of heat shock proteins HSP70-1A and HSP90β monitors the folding trajectory of the serotonin transporter. J Biol Chem. 2014 Oct 17; 289(42): 28987-29000.
- El Kasaby A, Just H, Malle E, et al. Mutations in the carboxyl-terminal SEC24 binding motif of the serotonin transporter impair folding of the transporter. J Biol Chem. 2010 Dec 10; 285(50): 39201-39210.
- Jacobs MT, Zhang YW, Campbell SD, et al. Ibogaine a noncompetitive inhibitor of serotonin transport acts by stabilizing the cytoplasm facing state of the transporter. J Biol Chem. 2007 Oct 5; 282(40): 29441-29447.
- Bulling S, Schicker K, Zhang YW, et al. The mechanistic basis for noncompetitive ibogaine inhibition of serotonin and dopamine transporters. J Biol Chem 2012 May 25; 287(22): 18524-18534.
- Korkhov VM, Holy M, Freissmuth M, et al. The conserved glutamate Glu136 in transmembrane domain 2 of the serotonin transporter is required for the conformational switch in the transport cycle. J Biol Chem. 2006 May 12; 281(19): 13439-13448.
- Sucic S, Dallinger S, Zdrazil B, et al. The N terminus of monoamine transporters is a lever required for the action of amphetamines. J Biol Chem. 2010 Apr 2; 285(14): 10924-10938.
- Koban F, El-Kasaby A, Häusler C, et al. A salt bridge linking the first intracellular loop with the C terminus facilitates the folding of the serotonin transporter. J Biol Chem. 2015 May 22; 290(21): 13263-78.
- Farhan H, Korkhov VM, Paulitschke V, et al. Two discontinuous segments in the carboxyl terminus are required for membrane targeting of the rat gamma-aminobutyric acid transporter 1 GAT1. J Biol Chem. 2004 Jul 2; 279(27): 28553-28563.
- Larsen MB, Fjorback AW, Wiborg O. The C terminus is critical for the functional expression of the human serotonin transporter. Biochemistry. 2006 Jan 31; 45(4): 1331-7.
- Sucic S, Bryan Lluka LJ. The role of the conserved GXXXRXG motif in the expression and function of the human norepinephrine transporter. Brain Res Mol Brain Res. 2002 Dec; 108(1-2): 40-50.
- Sucic S, Bryan Lluka LJ. Roles of transmembrane domain 2 and the first intracellular loop in human noradrenaline transporter function pharmacological and SCAM analysis. J Neurochem. 2005 Sep; 94(6): 1620-1630.
- Sucic S, Bryan Lluka LJ. Investigation of the functional roles of the MELAL and GQXXRXG motifs of the human noradrenaline transporter using cysteine mutants. Eur J Pharmacol. 2007 Feb 5; 556(1-3): 27-35.
- Reiterer V, Maier S, Sitte HH, et al. Sec24 and ARFGAP1 dependent trafficking of GABA transporter 1 is a prerequisite for correct axonal targeting. J Neurosci. 2008 Nov 19; 28(47): 12453-12464.
- Farhan H, Reiterer V, Kriz A, et al. Signal dependent export of GABA transporter 1 from the ER-Golgi intermediate compartment is specified by a C terminal motif. J Cell Sci. 2008 Mar 15; 121(Pt 6): 753-761.
- Sucic S, El Kasaby A, Kudlacek O, et al. The serotonin transporter is an exclusive client of the coat protein complex II COPII component SEC24C. J Biol Chem. 2011 May 6; 286(18): 16482-16490.
- Sucic S, Koban F, El Kasaby A, et al. Switching the clientele a lysine residing in the C terminus of the serotonin transporter specifies its preference for the coat protein complex II component SEC24C. J Biol Chem. 2013 Feb 22; 288(8): 5330-5341.
- Montgomery TR, Steinkellner T, Sucic S, et al. Axonal targeting of the serotonin transporter in cultured rat dorsal raphe neurons is specified by SEC24C dependent export from the endoplasmic reticulum. J Neurosci. 2014 Apr 30; 34(18): 6344-6351.
- Wu MN, Koh K, Yue Z, et al. A genetic screen for sleep and circadian mutants reveals mechanisms underlying regulation of sleep in Drosophila. Sleep. 2008 Apr; 31(4): 465-472.
- Kume K, Kume S, Park S K, et al. Dopamine is a regulator of arousal in the fruit fly. J. Neurosci. 2005 Aug 10; 25(32): 7377–7384.
- Kasture A, El Kasaby A, Szoelloesi D, et al. Functional rescue of a misfolded Drosophila melanogaster dopamine transporter mutant associated with a sleepless phenotype by pharmacological chaperones. J Biol Chem. 2016 Sept 30; 291: 20876-20890.
- Beerepoot P, Lam VM, Salahpour A.Pharmacological chaperones of the dopamine transporter rescue dopamine transporter deficiency syndrome mutations in heterologous cells. J Biol Chem. 2016 Oct 14; 291(42): 22053-22062.
- Lion François L, Cheillan D, Pitelet G, et al. High frequency of creatine deficiency syndromes in patients with unexplained mental retardation. Neurology. 2006 Nov 14; 67(9): 1713-1714.
- Rubenstein RC, Zeitlin PL. Sodium 4 phenylbutyrate downregulates Hsc70 implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol. 2000 Feb 1; 278: C259–C267.
- Fujiwara M, Yamamoto H, Miyagi T, et al. Effects of the chemical chaperone 4 phenylbutylate on the function of the serotonin transporter SERT expressed in COS 7 cells. J Pharmacol Sci. 2013 June 20; 122(2): 71-83.