
RIP140 in Stress Response of the Brain
Yu-Lung Lin1, Yi-Wei Li1, Li-Na Wei1*
Department of Pharmacology, University of Minnesota, Minneapolis, USA
Abstract
Receptor interacting protein 140 (RIP140), gene named Nuclear receptor interacting protein 1 (Nrip1), is a transcription co-regulator of numerous nuclear receptors and transcription factors that are important for various biological processes. RIP140 is highly expressed in various cell types of the brain, especially cortex and hippocampus. Increasingly, studies have begun to reveal its multiple functional roles in maintaining brain health. In particularly, there appears to be an intimate relationship between RIP140 and neurodegenerative diseases, such as reduced RIP140 expression in Alzheimer’s Disease (AD) postmortem brain and impaired cognitive functions in RIP140 knockout mice. The different functional roles of RIP140, mediated by distinct mechanisms, coordinately contribute to the execution of stress response of the brain to Endoplasmic Reticulum (ER) stress, heat shock (HS) stress, oxidative stress and psychological/behavioral stress. In this review, we describe the roles of RIP140 in three brain cell types (neurons, microglia, and astrocytes) stressed by pharmacological agents or behavioral manipulation. These results demonstrate physiological integration of various functional roles of RIP140 in different brain cells to facilitate survival and recovery from stress. The results also suggest a potential, preventive and/or therapeutic strategy by targeting RIP140 in managing neurodegenerative diseases.
Introduction
Receptor interacting protein RIP140 (RIP140, gene name Nrip1) is a versatile transcriptional co-regulator of numerous nuclear receptors and transcription factors1,2. RIP140 can function as a co-activator or co-repressor, and is detected in various tissues including brain, liver, adipose, muscle and immune cells. As a co-repressor, RIP140 recruits histone deacetylases (HDACs), C-terminus binding protein (CtBP) and other chromatin remodeling proteins to regulate gene expression2. In adipocyte, RIP140 suppresses genomic response to hormones such as thyroid hormone and retinoic acid3-5. Animal and cell culture studies have demonstrated that lipid accumulation, fatty acid oxidation, energy dissipation, and glucose uptake can be regulated by RIP140 in adipocyte6,7. In hepatocyte, RIP140 reduces phosphoenolpyruvate carboxykinase (PEPCK) levels to suppress gluconeogenesis by acting as a liver X receptor (LXR) co-repressor8. RIP140 also represses E2F transcription factor to affect tumor growth9,10. As a co-activator, RIP140 interacts with LXR to enhance the expression of sterol regulatory element-binding protein 1 (SREBP-1) and fatty acid synthase to promote lipogenesis in the liver8. In inflammatory (M1) macrophage, RIP140 acts as co-activator of NFκB to increase inflammatory response11.
In the brain, RIP140 is expressed in neurons, microglia and astrocytes, indicating extensive functional roles for RIP140 in maintaining brain health. Molecular studies have shown extensive protein modification and context-dependent distribution/transport of RIP140 under various physiological/pathological conditions (see section 2 below), supporting the complexity of RIP140’s multiple functional roles. Its ultimate role, on a system level, is most likely to reflect the collective actions of its multiple biological activities in various cell types rather than a simple cell-autonomous activity. To this end, this review focuses on RIP140’s functional roles in three major brain cell types, neurons, microglia and astrocytes. Established molecular mechanisms of RIP140’s action in the brain, mostly implicated in stress responses, are also reviewed.
Molecular features of RIP140
The first interesting molecular feature of RIP140 its extensively context-dependent post-translational modifications (PTMs) such as phosphorylation, methylation, acetylation, methylation, PLP conjugation, SUMOylation and ubiquitination2,12,13. These PTMs, elicited individually or in combination in response to various stimuli, modulate sub-cellular localization, stability and activity of RIP140. For instance, RIP140 undergoes specific arginine-methylation to initiate its nuclear-cytoplasmic export during adipocyte maturation/expansion14, which can also play out in stressed neurons (see later). In macrophages, in lipopolysaccharide induced inflammatory response, RIP140 is not only degraded via spleen protein kinase (Syk)-mediated tyrosine phosphorylation to tone down inflammatory response11 but also exported to the cytoplasm to activate STAT6 signaling and enhance M2 anti-inflammatory response15.
Secondly, RIP140 also exerts specific extra-nuclear activities in various cell types, besides its widely reported co-regulatory activities in gene transcription. In fat-accumulating, expanding adipocyte, RIP140 gradually translocate to the cytoplasm and interacts with AS160 (Akt substrate of 160 kDa), thereby reducing further uptake of glucose and insulin vesicles, which contributes insulin resistance7. In macrophage, RIP140 is exported to the cytoplasm of activated cell to enhance M2 polarization in anti-inflammatory stage15. The extra-nuclear activity of RIP140 is also observed in stressed neurons (see below).
Thirdly, in adult brain, RIP140 is detected in multiple cell types including neurons, microglia and astrocytes16-19. In neurons, it can also be distributed to the nucleus and/or cytoplasm, depending upon the states/conditions of the cells19,20. Further, its biological activity, protein stability and dynamics in neurons are context-dependent, which have been investigated in cultures and, to a lesser extent, in animals. Below we review the results of studies concerning RIP140 in these three brain cell types, and its physiological/pathological relevance to stresses in the brain. Table 1 lists reports that are related to RIP140’s activities and behavior in brain cells in vitro, as well as the implication of RIP140 in stress responses of the brain in vivo.
Table 1. The roles of RIP140 in various brain regions, cell subtypes and CNS diseases.
|
Author(s) and year |
Cell subtype or Brain area |
Stress/diseases model studied |
Role of RIP140 |
|
Chuang et al, 199766 |
GHFT1-5 pituitary progenitor cell line |
N/A |
Thyroid hormone receptor/ estrogen receptor related gene regulation |
|
Schaufele, 199967 |
GHFT1-5 pituitary progenitor cell line |
N/A |
Estrogen receptor related gene regulation |
|
Duclot et al, 201217 |
Cortex and hippocampus |
Alzheimer's disease and depression |
Cognitive learning and memory as well as stress response. |
|
Flaisher-Grinberg et al, 201450 |
Hypothalamus |
Forced swimming stress |
Emotional regulatory |
|
Feng et al, 201420 |
Neuron in hippocampus |
ER stress |
Neuron protection |
|
Feng et al, 201516 |
Astrocyte; cortex, hippocampus, and hypothalamus |
Behavioral stress for mice; ER stress for astrocyte |
Brain cholesterol homeostasis |
|
Blondrath et al, 201618 |
Hippocampus, cortex, cerebellum |
Alzheimer's disease
|
Regulation of gene involved in Ab generation |
|
Lin et al, 201719 |
Neuron in hippocampus |
Heat shock stress/oxidative stress |
Co-repressor of HSF1 and regulation of HSR |
RIP140 in neurons
Nuclear RIP140 regulates heat shock factor 1 (HSF1) and HSR in stressed neurons
HSR is a highly conserved mechanism that protects organisms from different stressors, such as HS, heavy metals, oxidative stress, and ER stress21,22. These stressors induce HSF1, the key transcription factor for HSR, which then dissociates from the chaperone complex and translocates into the nucleus. The active HSF1 forms phosphorylated trimers as an active transcription factor by binding to HS element (HSE) to promote the expression of HSR genes that are necessary for protein homeostasis and survival of stressed cells. In neurons derived from Huntington Disease (HD) patients’ brain, downregulating HSF1 induces the accumulation of toxic misfolded protein which is involved in neurodegeneration23,24. On the contrary, the presence of active HSF1 can prevent polyglutamine (polyQ) aggregate formation and attenuate neurodegeneration in cellular, mouse, worm, and ï¬y models25-27. The protective role of HSF1 activation is mediated by its induction of heat shock proteins (HSPs). Over-expression or pharmacological activation of HSPs is sufficient to suppress proteinopathic neurodegeneration21,28. All these studies demonstrate that HSF1-mediated HSR plays a crucial role in the development/progression of neurodegenerative diseases.
To this end, we have found that RIP140 is protective for hippocampal neurons in response to different stresses and cell death induction by acting as a co-repressor of HSF1 to down-regulate HSR in stressed hippocampal neuron19. In these neurons, HS rapidly (20 min at 41°C; 10 min at 43°C) induces HSF1 activation to elevate HSR. It appears that nuclear RIP140 can be associated with HSF1 as a corepressor when HSF1 enters the nucleus in stressed neurons. Following stress induction, rapid degradation of nuclear RIP140, which is otherwise associated with HSF1 as a corepressor, relieves HSF1 from repression and allows HSF1 to timely switch to an activator. Oxidative stress induced by sodium arsenite (As) at a concentration higher than 40 μM also induces RIP140 degradation and subsequent HSF1-mediated HSR. In primary and HT22 hippocampal neurons, silencing RIP140 can enhance HSR, rescue spine density, and protect neurons from As-induced oxidative stress19. In vivo experiments have shown that chronic as treatment causes significant hippocampal-dependent learning defects in mice. Chronic knockdown of RIP140 in hippocampus by lentivirus-delivery of RIP140-silencing RNA (shRIP140) efï¬ciently rescues the defects in As-treated mice, and dramatically increases HSR19. These animal experiments suggest that RIP140, possibly acting as a corepressor of HSF1, may function to safe guard neurons from accidental HSR under a normal condition; but in stressed neurons, early degradation of RIP140 may be crucial for neurons to activate HSF1 and subsequent HSR in order to adapt to the stressful conditions and survive. Thus, we propose a model that, timely reducing nuclear RIP140 level in neurons is important for the execution of HSR in stressed neurons, ultimately protecting the brain from stressful insults (Fig. 1).
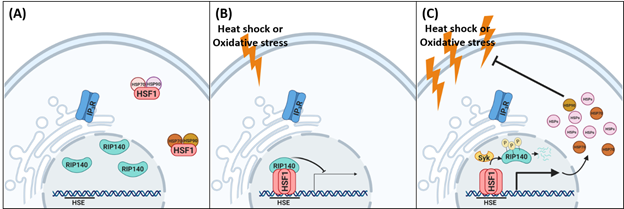
Figure 1: A scheme showing RIP140 modulating HSR under different stressful conditions in neuron. (A). In unstressed cells, HSF1 is localized in the cytosol and RIP140 is localized in nucleus. (B) During mild stress, HSF1 translocates to the nucleus to bind HSE and activate its chromatin targets; but RIP140 associates with HSF1 to repress (tone-down) HSF1’s transcriptional activity. (C) In severely stressed cells, Syk is activated to phosphorylate RIP140 for its degradation; the repression of HSF1 is thus relieved, enabling a full scale HSR that facilitates adaptation and recovery.
In this context, rapid RIP140 degradation provides a timely, cell-autonomous protective mechanism, enabling proper HSF1 activation in hippocampal neurons19. Molecularly, we have determined that HS and As-stress activates RIP140 degradation, mediated by a specific proteasome dependent protein degradation that involves Syk elicited phosphorylation on three specific Tyr residues (Tyr 364, 418, 436) of RIP140. Interesting, inflammatory stress in macrophages also induces RIP140 degradation via the same Syk-mediated Tyr-phosphorylation11, suggesting that Syk-mediated RIP140 degradation pathway may provide a common stress sensing mechanism in multiple cell types including neurons.
Cytoplasmic RIP140 protects neurons from ER stress via regulating IP3R
Eukaryotic cells employ a protein quality control important for cellular survival and function. Disrupting this system usually results in ER stress and the activation of unfolded protein response (UPR). ER stress is also closely related to Ca2+ homeostasis including its uptake and efflux through various transporters and ion channels on the ER membrane29. Depleting Ca2+ storage in ER by blocking Ca2+ pumps or disrupting Ca2+ release results in ER stress and cell death29. The control of intracellular Ca2+ via ER is contributed by two major ion channels/receptors - inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) and ryanodine receptors (RyRs)32. IP3R on the ER is the main Ca2+-release channel30,31. Importantly, overtly activating IP3Rs usually triggers pathological processes and apoptosis, and knockout IP3R1 enhances neuronal vulnerability to ER stress30. Moreover, stimulation of ER Ca2+ channels accelerates thapsigargin-induced ER calcium depletion and apoptosis and reducing Ca2+ release from ER by pharmacologically blocking IP3R-mediated Ca2+ release can attenuate ER stress and improved cell survival32. These studies demonstrate complicated regulation of IP3R in normal and stressed conditions.
We have found that, in neurons, ER stress inducers, such as thapsigargin (ER membrane Ca2+ pumps blocker), dithiothreitol (reducing agent), brefeldin A (collapse of the Golgi into ER), and amyloid beta (Aβ) all rapidly (within 15 min) induce nuclear RIP140 export and translocation to the cytoplasm20. In the cytoplasm, RIP140 binds specifically to the IP3R, but not RyRs, on ER membrane, thereby inhibiting IP3R-mediated Ca2+ release from ER and protecting hippocampal neuron from excitatory neurotoxicity and cell death20. We have further employed an animal model of ER stress (intraperitoneal injection of Tg) to validate the functional role for RIP140 in vivo33. In this study, RIP140 was knocked down in hippocampal CA1 region, resulting in enhanced ER stress and apoptosis markers following Tg treatment, as compared to the control animals (without RIP140 silencing)20. These results together support a functional role for RIP140 in neuroprotection against ER stress in vitro and in vivo.
At the molecular level, we have determined that the RD4 domain, residing at the carboxyl terminus of RIP140, directly interacts with the C-terminal gate-keeper domain of IP3R, which disrupts the ‘’head-tail’’ interaction of IP3R necessary for its activation, thereby inhibiting channel open and Ca2+ release. Interestingly, introducing the RD4 fragment of RIP140 into hippocampal neurons is sufficient to decrease the formation of endogenous RIP140/IP3R complex and reduce Ca2+ release20, suggesting a potential therapeutic strategy by targeting RIP140 in managing neuronal diseases associated with ER stress (Fig. 2).
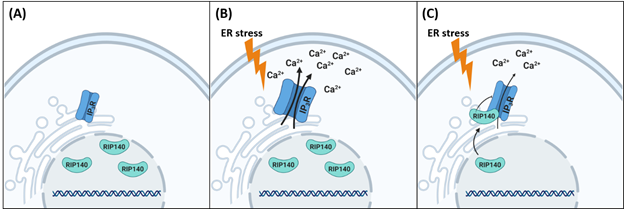
Figure 2: A scheme showing RIP140 modulating ER (cytosolic) stress via interacting with IP3R. (A). In unstressed cells, RIP140 is localized in nucleus. (B) During ER stress, IP3Rs are activated and Ca2+ is released from ER, which results in cell apoptosis. (C) To facilitate cell survival, RIP140 is rapidly translocated to the cytoplasm, specifically interacting with IP3R on the ER membrane, to reduce over-stimulation of IP3R-mediated Ca2+ release, thereby protecting neuron from ER stress-induced cell death.
These studies illustrate at least two functional roles for RIP140 in protecting neurons from ER stress, mediated by its nuclear form (interacting with HSF1) and cytoplasmic form (interacting with IP3R). These neuron cell-autonomous protective mechanisms can contribute to the brain health. Their dysfunction may contribute to certain stress-related brain diseases as implicated in clinical data that show correlation with abnormal expression of RIP14018.
RIP140 in microglia
The Immune system and the central nervous system (CNS) interact closely to constitute the neuro-immune axis, which plays an important role in the development and homeostasis of the nervous system34,35. Emerging studies have demonstrated that maintaining the crosstalk within this axis is pivotal to various neurophysiological processes36,37. Due to physical blood-brain barrier, immune responses within the CNS are different from those in the periphery including the peripheral nervous system (PNS)38,39. In the brain, tissue-resident macrophages, microglia cells, are one of the essential cell populations involved in defensive immune responses including sensing environmental changes and neuroprotection34. Therefore, neuro-immune dysregulation can cause microglia cells to lose neuroprotective functions and trigger neurotoxicity, leading to various neurological diseases40,41. For instance, during neuronal inflammation, microglia cells could activate astrocytes, causing amplification and sustention of inflammatory status and further leading to neurotoxicity42-44. In AD, microglia cells also play an important role in cleaning Aβ deposition through phagocytosis and endocytosis45. In brain development, microglia are critical for synapse pruning; importantly, they can be reactivated in disease conditions including AD46-49.
In our lab, using a monocyte-specific RIP140 knockdown (MfRIP140KD) mouse model11 where microglia RIP140 level is also reduced, we have found that MfRIP140KD mice have increased anxiety- and depressive-like behaviors50. Interestingly, in MfRIP140KD mice where RIP140 expression is reduced in microglia, there appears to be a significant reduction in neuropeptide Y production in astrocyte. It is tempting to speculate that augmenting RIP140 levels in microglia, and thus brain inflammatory status, can also affect brain neurochemistry and contribute to neurological disorders. To this end, as increasingly recognized, microglia dysregulation can be highly associated with neurological diseases especially those related to chronic stress in the brain. In this context, RIP140’s action in microglia can potentially contribute to stress response in the brain on a system level.
RIP140 in astrocyte
Clinical and basic studies both have demonstrated that stressful experiences can result in psychological and neurological disorders including depression51,52. Forced swimming stress (FSS) test is the most popular test for depression-like behaviors in rodents, and is wildly used as an antidepressant screening tool in rodents modeling human psychological stress53.
MfRIP140KD mice show a significant increase in immobility when challenged with FSS, indicating a role for RIP140 in depressive like stress response17. While FSS can trigger a wide range of changes such as the sympathetic nervous system54, the HPA axis (increased the stress hormone, corticosterone)54,55, and neurotransmitters such as dopamine, serotonin, and GABA in the brain56,57; of interest is that FSS also significantly reduces RIP140 level in the brain and increases its cholesterol content, which is associated with increased ER stress markers such as Bip in mouse brain16. Using primary astrocyte cell cultures as experimental systems, we have found that ER stress directly inhibits RIP140 mRNA expression via increasing microRNA 33 (miR33)16 which targets the mRNA’s 3’-untranslated region of RIP14058. Further, lowering RIP140 levels in astrocytes disrupts the expression of cholesterol metabolic genes and accelerates cholesterol exportation from astrocytes and uptake by neurons16. Presumably, these are attributable to the co-regulator (such as for the expression of cholesterol metabolizing genes) function of nuclear RIP140 in astrocyte.
While preliminary, these studies indicate that RIP140 in astrocyte can affect brain cholesterol homeostasis, which may also modulate depressive-like stress in animals, and, probably, psychological stress-related conditions and other neurological disorders like AD in humans. Studies have also indicated that RIP140 can be involved in Aβ plaque formation18,58,61. To this end, Apolipoprotein E4 has been shown as a key player in cholesterol regulation and Aβ plaque formation, and is a known risk factor of AD59,60. It would be interesting to examine if and how RIP140 may be related to the regulation ApoE. Rigorous mechanistic studies of RIP140 action in astrocyte, in vitro and in vivo, await to be carried out.
Discussion and Perspectives
Because of the complicated PTMs of RIP140, which affects its protein localization, stability and functionality, and the extremely context-dependent nature of RIP140’s activity/behavior, it requires extensive studies to carefully dissect and integrate the physiological and/or pathological pathways in specific brain cell types/circuitries where RIP140 plays roles in the face of stressful condition. It is also important to take into account the sequence of these events happening in a specific physiological or pathological processes. For example, ER stress and inflammation both can induce oxidative stress and HSR. However, ER stress induced by thapsigargin, dithiothreitol, and brefeldin A promotes rapid RIP140 translocation to cytoplasm in hippocampal neurons where it is required for toning down the extent of ER stress-induced IP3R activity, thus protecting cells from apoptosis20. But HS, As-induced oxidative stress in hippocampal neurons can rapidly trigger nuclear RIP140 degradation, allowing HSF1 activation and thus execution of HSR program19. These represent neuron cell-autonomous protective mechanisms in the face of stress, mediated by both the nuclear and the cytoplasmic forms of RIP140. In astrocyte, ER stress reduces RIP140 mRNA and protein level16, which can alter brain neurochemistry and ultimately contributes to the modulation of brain health. In microglia, RIP140 modulates inflammatory response for both inflammatory and anti-inflammatory phases. This also is crucial to brain function and the maintenance of brain health. Given these multiple functions of RIP140 in various brain cells, it is important to understand the precise mechanism regulating RIP140 behavior/activity in these various brain cell types, and to what extent RIP140 is involved in these various processes in the context of whole brain. How these potentially synergistic or additive functional roles of RIP140 in various brain cells may integrate in the face of a specific type of stress, and whether any malfunction of RIP140 in any of these cells may contribute to the pathological processes of brain diseases are important questions to be further examined.
In the context of whole animal, the role of RIP140 specifically attributable to its actions in the brain has just begun to be understood. The facts that RIP140 is expressed in multiple cell types of the brain and that it can function, differently, in the nucleus vs. the cytoplasm would indicate very complicated, multiple mechanisms of action. Accumulated data have mostly revealed its ultimate action in modulating brain stress response. This can be mediated by its cell autonomous action, such as in directly modulating neuronal stress, or by its action in non-neuronal cells like microglia and/or astrocyte that ultimately contribute to the overall health of the brain. In adult brain, RIP140 is most prominently detected in the cortex and hippocampus18,62. This is also consistent with the finding that whole body RIP140 knockout mice indeed exhibit impaired cognitive function including deficits in hippocampus-dependent memory and learning, as well as an increased response to FSS challenges17. More interestingly, RIP140 expression in the brain is reduced during aging63 and in AD postmortem brains18. In RIP140 transgenic (Nrip1 knockout or over-expressing human RIP140) mice and cell lines, it has been found that RIP140 can regulate the expression of AD related genes, including β-site APP cleaving enzyme 1 (BACE1) and glycogen synthase kinase-3 (GSK3)18. Finally, the human Nrip1 gene is located on chromosome 2164 (an extra copy would cause Down’s syndrome), and in the hippocampus of Down’s syndrome patients, RIP140 level is elevated65. All these experimental cell and animal studies, as well as clinical data, indicate that RIP140 can be intimately related to the development and/or progression of certain brain disorders, most notably stress-related neurodegenerative diseases such as AD and developmental disorders like Down’s syndrome.
Many neurodegenerative diseases involve inflammation and protein misfolding and/or aggregation, such as Aβ and tau in AD, α-synuclein in Parkinson’s disease, Huntingtin in HD, and superoxide dismutase in Amyotrophic lateral sclerosis. As described above, RIP140 exerts protective functions to attenuate, or adapt to, the stress condition in stressed cells, depending upon various specific signaling pathways (which can be triggered by environmental factors and/or cellular state that elicits PTMs). Therefore, it may be feasible to identify specific compounds that can trigger specific RIP140 translocation and/or degradation, thereby activating specific protective mechanism. Physiologically, stress responses are essential but proper and timely execution of different stress responses must be tightly regulated and coordinated in order to achieve the ultimate goal of animal survival and maintaining health. Given the high specificity of RIP140’s functions and relevant signaling pathways, RIP140 should provide a highly potential and selective target for designing therapeutic strategy in the future. This requires careful dissection of the complicated and integrated functional roles of various forms of RIP140 in particular cell types in future studies.
Acknowledgement
This work is supported by NIH grants DK54733, DK60521, and the Dean’s Commitment and the Distinguished McKnight Professorship of University of Minnesota to LNW. A Conflict of Interest statement is included in the main manuscript file and appears before the reference listing.
References
- Wei,LN. Retinoids and receptor interacting protein 140 (RIP140) in gene regulation. Current medicinal chemistry. 2004; 11: 1527-1532.
- Mostaqul Huq MD, Gupta P, Wei LN. Post-translational modifications of nuclear co-repressor RIP140: a therapeutic target for metabolic diseases. Current medicinal chemistry. 2008; 15: 386-392.
- Gupta P, Park SW, Farooqui M, et al. Orphan nuclear receptor TR2, a mediator of preadipocyte proliferation, is differentially regulated by RA through exchange of coactivator PCAF with corepressor RIP140 on a platform molecule GRIP1. Nucleic Acids Res. 2007; 35: 2269-2282, doi:10.1093/nar/gkl1147.
- Park SW, Huang WH, Persaud SD, et al. RIP140 in thyroid hormone-repression and chromatin remodeling of Crabp1 gene during adipocyte differentiation. Nucleic Acids Res. 2009; 37: 7085-7094, doi:10.1093/nar/gkp780.
- Persaud SD, Huang WH, Park SW, et al. Gene repressive activity of RIP140 through direct interaction with CDK8. Mol Endocrinol. 2011; 25: 1689-1698, doi:10.1210/me.2011-1072.
- Leonardsson G, Steel JH, Christian M, et al. Nuclear receptor corepressor RIP140 regulates fat accumulation. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101: 8437-8442, doi:10.1073/pnas.0401013101.
- Ho PC, Lin YW, Tsui YC, et al. A negative regulatory pathway of GLUT4 trafficking in adipocyte: new function of RIP140 in the cytoplasm via AS160. Cell metabolism. 2009; 10: 516-523, doi:10.1016/j.cmet.2009.09.012.
- Herzog B, Hallberg M, Seth A, et al. The nuclear receptor cofactor, receptor-interacting protein 140, is required for the regulation of hepatic lipid and glucose metabolism by liver X receptor. Mol Endocrinol. 2007; 21: 2687-2697, doi:10.1210/me.2007-0213.
- Docquier A, Harmand PO, Fritsch S, et al. The transcriptional coregulator RIP140 represses E2F1 activity and discriminates breast cancer subtypes. Clin Cancer Res. 2010; 16: 2959-2970, doi:10.1158/1078-0432.CCR-09-3153.
- Lapierre M, Docquier A, Castet-Nicolas A, et al. The emerging role of the transcriptional coregulator RIP140 in solid tumors. Biochim Biophys Acta. 2015; 1856: 144-150, doi:10.1016/j.bbcan.2015.06.006.
- Ho PC, Tsui YC, Feng X, et al. NF-kappaB-mediated degradation of the coactivator RIP140 regulates inflammatory responses and contributes to endotoxin tolerance. Nature immunology. 2012; 13: 379-386, doi:10.1038/ni.2238.
- Wu CY, Persaud SD, Wei LN. Retinoic Acid Induces Ubiquitination-Resistant RIP140/LSD1 Complex to Fine-Tune Pax6 Gene in Neuronal Differentiation. Stem Cells. 2016; 34: 114-123, doi:10.1002/stem.2190.
- Huq MD, Ha SG, Barcelona H, et al. Lysine methylation of nuclear co-repressor receptor interacting protein 140. Journal of proteome research. 2009; 8: 1156-1167, doi:10.1021/pr800569c.
- Gupta P, Ho PC, Huq MD, et al. PKCepsilon stimulated arginine methylation of RIP140 for its nuclear-cytoplasmic export in adipocyte differentiation. PloS one. 2008; 3: e2658, doi:10.1371/journal.pone.0002658.
- Lin YW1, Lee B, Liu PS, et al. Receptor-Interacting Protein 140 Orchestrates the Dynamics of Macrophage M1/M2 Polarization. Journal of innate immunity. 2016; 8: 97-107, doi:10.1159/000433539.
- Feng X, Lin YL, Wei LN. Behavioral stress reduces RIP140 expression in astrocyte and increases brain lipid accumulation. Brain behavior and immunity. 2015; 46: 270-279, doi:10.1016/j.bbi.2015.02.008.
- Duclot F, Lapierre M, Fritsch S, et al. Cognitive impairments in adult mice with constitutive inactivation of RIP140 gene expression. Genes Brain Behav. 2012; 11: 69-78, doi:10.1111/j.1601-183X.2011.00731.x.
- Blondrath K, Steel JH, Katsouri L, et al. The nuclear cofactor receptor interacting protein-140 (RIP140) regulates the expression of genes involved in Abeta generation. Neurobiol Aging. 2016; 47: 180-191, doi:10.1016/j.neurobiolaging.2016.08.003.
- Lin YL, Tsai HC, Liu PY, et al. Receptor-interacting protein 140 as a co-repressor of Heat Shock Factor 1 regulates neuronal stress response. Cell death & disease. 2017; 8: 3203, doi:10.1038/s41419-017-0008-5.
- Feng X, Krogh KA, Wu CY, et al. Receptor-interacting protein 140 attenuates endoplasmic reticulum stress in neurons and protects against cell death. Nature communications. 2014; 5: 4487, doi:10.1038/ncomms5487.
- Bose S, Cho J. Targeting chaperones, heat shock factor-1, and unfolded protein response: Promising therapeutic approaches for neurodegenerative disorders. Ageing research reviews. 2016. doi:10.1016/j.arr.2016.09.004.
- Velichko AK, Markova EN, Petrova NV, et al. Mechanisms of heat shock response in mammals. Cellular and molecular life sciences : CMLS. 2013; 70: 4229-4241, doi:10.1007/s00018-013-1348-7.
- Gomez-Pastor R, Burchfiel ET, Neef DW, et al. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington's disease. Nature communications. 2017; 8: 14405, doi:10.1038/ncomms14405.
- Kim E, Wang B, Sastry N, et al. NEDD4-mediated HSF1 degradation underlies alpha-synucleinopathy. Human molecular genetics. 2016; 25: 211-222, doi:10.1093/hmg/ddv445.
- Fujimoto M, Takaki E, Hayashi T, et al. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. The Journal of biological chemistry. 2005; 280: 34908-34916, doi:10.1074/jbc.M506288200.
- Morley JF, Brignull HR, Weyers JJ, et al. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99: 10417-10422, doi:10.1073/pnas.152161099.
- Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010; 8: e1000291, doi:10.1371/journal.pbio.1000291.
- Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harbor symposia on quantitative biology. 2011; 76: 91-99, doi:10.1101/sqb.2012.76.010637.
- Yates D. Neurodegenerative disease: The stress of misfolding. Nature reviews. Drug discovery. 2012; 11: 352-353, doi:10.1038/nrd3731.
- Higo T, Hamada K, Hisatsune C, et al. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron. 2010; 68: 865-878, doi:10.1016/j.neuron.2010.11.010.
- Green DR, Wang R. Calcium and energy: making the cake and eating it too? Cell. 2010; 142: 200-202, doi:10.1016/j.cell.2010.07.007.
- Luciani DS, Gwiazda KS, Yang TL, et al. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes. 2009; 58: 422-432, doi:10.2337/db07-1762.
- Hetz C, Bernasconi P, Fisher J, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006; 312: 572-576, doi:10.1126/science.1123480.
- Norris GT, Kipnis J. Immune cells and CNS physiology: Microglia and beyond. J Exp Med. 2019; 216: 60-70, doi:10.1084/jem.20180199.
- Morimoto K, Nakajima K. Role of the Immune System in the Development of the Central Nervous System. Front Neurosci. 2019; 13: 916, doi:10.3389/fnins.2019.00916.
- Williams JL, Holman DW, Klein RS. Chemokines in the balance: maintenance of homeostasis and protection at CNS barriers. Front Cell Neurosci. 2014; 8: 154, doi:10.3389/fncel.2014.00154.
- Veiga-Fernandes H, Artis D. Neuronal-immune system cross-talk in homeostasis. Science. 2018; 359: 1465-1466, doi:10.1126/science.aap9598.
- Louveau A, Harris TH, Kipnis J. Revisiting the Mechanisms of CNS Immune Privilege. Trends in immunology. 2015; 36: 569-577, doi:10.1016/j.it.2015.08.006.
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends in immunology. 2007; 28: 12-18, doi:10.1016/j.it.2006.11.004.
- Hickman S, Izzy S, Sen P, et al. Microglia in neurodegeneration. Nat Neurosci. 2018; 21: 1359-1369, doi:10.1038/s41593-018-0242-x.
- Bohlen CJ, Friedman BA, Dejanovic B, et al. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu Rev Genet. 2019; 53: 263-288, doi:10.1146/annurev-genet-112618-043515.
- Tjalkens RB, Popichak KA, Kirkley KA. Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv Neurobiol. 2017; 18: 159-181, doi:10.1007/978-3-319-60189-2_8 (2017).
- Kirkley KS, Popichak KA, Afzali MF. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. Journal of neuroinflammation. 2017; 14: 99, doi:10.1186/s12974-017-0871-0.
- Liddelow SA, Guttenplan KA, Clarke LE et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017; 541: 481-487, doi:10.1038/nature21029.
- Frenkel D, Wilkinson K, Zhao L, et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nature communications. 2013; 4: 2030, doi:10.1038/ncomms3030.
- Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011; 333: 1456-1458, doi:10.1126/science.1202529.
- Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012; 74: 691-705, doi:10.1016/j.neuron.2012.03.026.
- Reichwald J1, Danner S, Wiederhold KH, et al. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. Journal of neuroinflammation. 2009; 6: 35, doi:10.1186/1742-2094-6-35.
- Stephan AH, Madison DV, Mateos JM, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013; 33: 13460-13474, doi:10.1523/JNEUROSCI.1333-13.2013.
- Flaisher-Grinberg S, Tsai HC, Feng X, et al. Emotional regulatory function of receptor interacting protein 140 revealed in the ventromedial hypothalamus. Brain behavior and immunity. 2014; 40: 226-234, doi:10.1016/j.bbi.2014.03.019.
- Juruena MF, Eror F, Cleare AJ, et al. The Role of Early Life Stress in HPA Axis and Anxiety. Advances in experimental medicine and biology. 2020; 1191: 141-153, doi:10.1007/978-981-32-9705-0_9.
- Moreno-Rius J. The cerebellum under stress. Frontiers in neuroendocrinology. 2019; 54: 100774, doi:10.1016/j.yfrne.2019.100774.
- Yan K, Chen YB, Wu JR, et al. Current Rapid-Onset Antidepressants and Related Animal Models. Current pharmaceutical design. 2018; 24: 2564-2572, doi:10.2174/1381612824666180727115222.
- Balkan B, Keser A, Gozen O, et al. Forced swim stress elicits region-specific changes in CART expression in the stress axis and stress regulatory brain areas. Brain research. 2012; 1432: 56-65, doi:10.1016/j.brainres.2011.11.006.
- Armario A, Gavalda A, Marti J. Comparison of the behavioural and endocrine response to forced swimming stress in five inbred strains of rats. Psychoneuroendocrinology. 1995; 20: 879-890, doi:10.1016/0306-4530(95)00018-6.
- Briones-Aranda A, Rocha L, Picazo O. Influence of forced swimming stress on 5-HT1A receptors and serotonin levels in mouse brain. Progress in neuro-psychopharmacology & biological psychiatry. 2005; 29: 275-281, doi:10.1016/j.pnpbp.2004.11.011.
- Briones-Aranda A, Rocha L, Picazo O. Alterations in GABAergic function following forced swimming stress. Pharmacology, biochemistry, and behavior. 2005; 80: 463-470, doi:10.1016/j.pbb.2005.01.002.
- Ho PC, Chang KC, Chuang YS, et al. Cholesterol regulation of receptor-interacting protein 140 via microRNA-33 in inflammatory cytokine production. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011; 25: 1758-1766, doi:10.1096/fj.10-179267.
- Williams T, Borchelt DR, Chakrabarty P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer's disease. Mol Neurodegener. 2020; 15: 8, doi:10.1186/s13024-020-0358-9.
- Jeong W, Lee H, Cho S, et al. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer's Disease. Mol Cells. 2019; 42: 739-746, doi:10.14348/molcells.2019.0200.
- Lin YW, Liu PS, Adhikari N, et al. RIP140 contributes to foam cell formation and atherosclerosis by regulating cholesterol homeostasis in macrophages. Journal of molecular and cellular cardiology. 2015; 79: 287-294, doi:10.1016/j.yjmcc.2014.12.009.
- Zhang XR, Zeng CM, DU JB, et al. [Expression profile of receptor-interacting protein 140 in the brain: experiment with prenatal and postnatal mice]. Zhonghua yi xue za zhi. 2007; 87: 3152-3154.
- Ghosh S, Thakur MK. Tissue-specific expression of receptor-interacting protein in aging mouse. Age (Dordr). 2008; 30: 237-243, doi:10.1007/s11357-008-9062-3.
- Katsanis N1, Ives JH, Groet J, et al. Localisation of receptor interacting protein 140 (RIP140) within 100 kb of D21S13 on 21q11, a gene-poor region of the human genome. Hum Genet. 1998; 102: 221-223, doi:10.1007/s004390050682.
- Gardiner K. Transcriptional dysregulation in Down syndrome: predictions for altered protein complex stoichiometries and post-translational modifications, and consequences for learning/behavior genes ELK, CREB, and the estrogen and glucocorticoid receptors. Behav Genet. 2006; 36: 439-453, doi:10.1007/s10519-006-9051-1.
- Chuang FM, West BL, Baxter JD, et al. Activities in Pit-1 determine whether receptor interacting protein 140 activates or inhibits Pit-1/nuclear receptor transcriptional synergy. Mol Endocrinol. 1997; 11: 1332-1341, doi:10.1210/mend.11.9.9978.
- Schaufele F. Regulation of estrogen receptor activation of the prolactin enhancer/promoter by antagonistic activation function-2-interacting proteins. Mol Endocrinol. 1999; 13: 935-945, doi:10.1210/mend.13.6.0298.